
Abstract
Background
Renin-angiotensin system (RAS) blockade during nephrogenesis causes a broad range of renal mal-development. Here, we hypothesized that disruption of renal lymphangiogenesis may contribute to tubulointerstitial alterations after RAS blockade during kidney maturation.
Methods
Newborn rat pups were treated with enalapril (30 mg/kg/day) or vehicle for 7 days after birth. Lymphangiogenesis was assessed via immunostaining and/or immunoblots for vascular endothelial growth factor (VEGF)-C, VEGF receptor (VEGFR)−3, Podoplanin, and Ki-67. The intrarenal expression of fibroblast growth factor (FGF)−1, FGF-2, FGF receptor (R)−1, α-smooth muscle actin (α-SMA), and fibroblast-specific protein (FSP)−1 was also determined. Sirius Red staining was performed to evaluate interstitial collagen deposition.
Results
On postnatal day 8, renal lymphangiogenesis was disrupted by neonatal enalapril treatment. The expression of podoplanin and Ki-67 decreased in enalapril-treated kidneys. While the expression of VEGF-C was decreased, the levels of VEGFR-3 receptor increased following enalapril treatment. Enalapril treatment also reduced the renal expression of FGF-1, FGF-2, and FGFR-1. Enalapril-treated kidneys exhibited profibrogenic properties with increased expression of α-SMA and FSP-1 and enhanced deposition of interstitial collagen.
Conclusion
Enalapril treatment during postnatal renal maturation can disrupt renal lymphangiogenesis along with tubulointerstitial changes, which may result in a pro-fibrotic environment in the developing rat kidney.
Similar content being viewed by others


Introduction
Lymphatic vessels play key roles in tissue fluid homeostasis, transport of macromolecules, and immune surveillance.1 Lymphatic growth, maturation, and regression are important in tissue remodeling during embryonic development, tissue repair, and inflammation.2 The kidney has well-developed lymphatic structures, and the renal lymphatic vasculature and lymphangiogenesis play a key role in kidney diseases, including renal fibrosis.3 Despite similarities between pathological lymphangiogenesis and normal physiology,4 the development of lymphatic vessels during nephrogenesis remains insufficiently investigated.
The growth and development of lymphatic vessels are coordinated by a large set of genes including those encoding the vascular endothelial growth factor receptor (VEGFR)−3 and its main ligand VEGF-C.5 VEGF-C is an important chemotactic and survival factor during lymphangiogenesis since homozygous deletion of VEGF-C induces complete lymphatic loss in embryos.6 Podoplanin, a transmembrane glycoprotein, promotes lymphatic cell adhesion and regulates the lymphatic vasculature. Gene targeting of podoplanin in mice induced dilated and malfunctioning lymphatic vessels.7 Ki-67 is a nuclear protein detected in all the proliferating cells during the cell cycle. More than 80% of lymphatic endothelial cells during the process of lymphangiogenesis expressed Ki-67 in developing rat kidneys.4 The fibroblast growth factor (FGF) family regulates many developmental processes, such as cell proliferation, migration, and differentiation.8 FGFs act via binding to high-affinity tyrosine kinase FGF receptors (FGFRs) denoted FGFR-1 through FGFR-4.9 FGF family of ligands and receptors are well-known as essential modulators of renal organogenesis while FGF-2 and its receptor FGFR-1 are also recognized as lymphangiogenic growth factors and mediators.1,10 In regard to adult kidney diseases, studies have implicated FGF-1 and FGF-2 in renal inflammation and fibrogenesis.11,12 However, many of these aspects in renal pathology remain to be elucidated.
Epithelial-to-mesenchymal transition (EMT) and its reverse process, mesenchymal-to-epithelial transition (MET), are implicated in various disease states as well as organ development.10 Cellular plasticity leads to altered cell morphology and interactions with the surrounding extracellular environment and adjacent cells.10 In the context of renal diseases, perturbation of homeostatic crosstalk between tubular epithelial cells and interstitial fibroblasts (or mesenchymal cells) leads to complete reorganization of the tubulointerstitial compartment.13 Nephron induction and maturation occurs continuously in the cortex of the developing kidney. In rodents, this process continues until postnatal day 10.14 The developing kidney strongly expresses all the components of the renin-angiotensin system (RAS) early in renal development and RAS blockade during nephrogenesis results in a broad range of renal mal-development.15,16 Administration of enalapril or losartan to newborn rats resulted in persistent renal lesions at different ages, ranging from 4 to 30 weeks after birth, including an increase in the interstitial volume along with fibrosis.16 We showed previously that RAS inhibition induced renal EMT, as well as capillary rarefaction in developing rat kidneys.17,18 Here, we hypothesized that disruption of renal lymphangiogenesis may also contribute to tubulointerstitial alterations after RAS blockade during kidney maturation. While angiotensin II plays an important role in kidney development and growth, renal lymphangiogenesis in association with RAS has yet to be characterized. Besides, molecular mechanisms that govern the renal histological lesions via RAS inhibition in immature kidneys are poorly understood. In this study we sought to investigate the effects of RAS inhibition on renal lymphangiogenesis together with the expression of renal MET/EMT modulators in the developing rat kidney.
Methods
Animal preparation
The animal protocol was reviewed and accepted by the Animal Experimentation Ethics Committee of Korea University and was performed under the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Twenty litters of Sprague–Dawley rats were treated with enalapril (n = 12, 30 mg/kg/day; Sigma Chemical Co., St. Louis, MO) dissolved in 0.9% saline or vehicle (n = 8, normal saline) via an orogastric tube daily within 24 h of birth. On day 8, each animal was euthanized with intraperitoneal pentobarbital sodium (50 mg/kg) and the respective kidneys were harvested and processed as described earlier.17
Histological and immunohistochemical (IHC) examinations
To assess cell proliferation, IHC staining of the nuclear antigen Ki-67 was conducted. The Ki-67 positive cells were assessed using the avidin-biotin immunoperoxidase method (Vectastain ABC kit). The primary antibodies included the monoclonal anti-mouse Ki-67 antibody (1:100 dilutions, DAKO, Glostrup, Denmark) in the study group and phosphate buffered saline (PBS) in the negative control group. Each slide was cross-stained with 0.5% methyl green solution (Trevigen). The number of Ki-67 positive cells was calculated by counting at least 20 grid fields (25 × 25 μm2) and obtaining the average results. Microscopic identification ( × 400) of tubules, interstitium and glomeruli in the cortex was performed using a double-blind method for each of the eight to twelve rats. The count was done randomly throughout all of the fields analyzed.
For IHC analyses, 8 to 12 kidneys in each group were selected for representative immunohistochemistry of podoplanin, VEGF-C, VEGFR-3, FGF-1, FGF-2, FGFR-1, fibroblast-specific protein (FSP)−1, and α-smooth muscle actin (α-SMA), using an avidin-biotin immunoperoxidase method (Vectastain ABC kit, Burlingame, CA). IHC study was performed as described previously.17 Primary antibodies against podoplanin (1:100), VEGF-C (1:100), VEGFR-3 (1:100), FGF-1 (1:200), FGF-2 (1:200), FGFR-1 (1:200), FSP-1 (1:250), and α-SMA (1:800) were used. All the primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The primary antibody was substituted with PBS as a negative control. The secondary antibodies were peroxidase-conjugated anti-rabbit IgG antibodies (1:1000; Millipore, Temecula, CA).
To detect interstitial collagen deposition, collagen content was measured by Sirius Red staining. After de-paraffinization, the kidney sections were hydrated and incubated in picrosirius red solution (1% Sirius Red in saturated picric acid) for 18 h, followed by 0.01 N HCl treatment for 2 min and dehydration. Eight to twelve kidneys in each group were examined using a double-blind method. For quantitative evaluation of the degree of fibrosis, 10 fields per section were selected randomly and the positive red-stained areas in the renal cortex were analyzed (magnification, × 200). Each analyzed positive area was expressed as a percentage of the total area within the field, using computerized image analysis software (Image Pro 5.1, Media Cybernetics Inc., MD).
Western blotting analysis
Protein extractions and Western blotting were performed as described earlier.17 Membranes were incubated for 18 h at 4 °C with primary antibodies against VEGF-C (dilution 1:200), VEGFR-3 (1:200), FGF-1 (1:500), FGF-2 (1:400), FGFR-1(1:500), fibrinogen (1:500), intercellular adhesion molecule (ICAM)−1 (1:400), toll-like receptor (TLR)−2 (1:400), and TLR-4 (1:400). All primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies were then applied for 60 min with anti-rabbit IgG (Ap132p, 1:1000; Millipore, Temecula, CA), anti-mouse IgG (1:1000; Cell Signaling Technology, Danvers, MA), or anti-goat IgG (HAF-109; R&D Systems, Minneapolis, MN) at room temperature. To control for equal loading, ß-actin (1:1000; Cell Signaling Technology, Danvers, MA), and anti-mouse IgG conjugated horseradish peroxidase (1:1000; Cell Signaling Technology, Danvers, MA) were used as primary and secondary antibodies using the same method described above. X-rays were scanned using the Epson GT-9500 (Seiko Corp, Nagano, Japan), and the results were quantified by a computerized densitometer (Image PC Alpha 9; National Institutes of Health, Bethesda, MD).
Statistical analysis
Data are presented as mean ± SEM. Differences between the groups were analyzed with the t-test for unpaired normally distributed data, and with Mann–Whitney rank sum test for data not normally distributed. A p-value <0.05 was considered significant. Statistical analyses were performed with SPSS ver. 16.0 for Windows software (SPSS Inc., Chicago, IL).
Results
Renal cell proliferation and lymphangiogenic markers
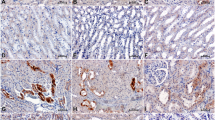
We estimated the intrarenal expression of the most extensively studied lymphatic vascular markers, podoplanin, VEGF-C, VEGFR-3, and cell proliferation marker Ki-67.4 IHC staining showed reduced expression of Ki-67 (Fig. 1a–c), VEGF-C (Fig. 1d–f) and podoplanin (Fig. 1j and k), in the enalapril-treated neonatal rat kidneys, compared with the controls. The number of Ki-67-positive nuclei in renal cortex was lower in the enalapril-treated group than in the control kidneys (P < 0.05) (Fig. 1c). VEGF-C, a well-known lymphangiogenic growth factor,19 was frequently detected in tubular epithelial cells in the normal control kidneys (Fig. 1d). However, it was rarely detected in the tubular cells in the enalapril-treated kidneys (Fig. 1e). The immunoblots additionally showed that the VEGF-C/β-actin protein expression decreased significantly in the enalapril-treated group (P < 0.05) (Fig. 1f). Podoplanin-positive lymphatic vessels were encountered mainly in the corticomedullary areas of normal kidneys on postnatal day 8 (Fig. 1j), whereas they were weakly detected in the kidneys of enalapril-treated rats (Fig. 1k). In contrast, VEGFR-3, a receptor for VEGF-C, was highly localized in tubular epithelial cells of the enalapril-treated kidneys (Fig. 1 h), while it was weakly observed in the normal control rat kidneys (Fig. 1g). The VEGFR-3/β-actin protein expression increased significantly in the enalapril-treated group, in comparison to control rats (P < 0.05) (Fig. 1i).

Intrarenal lymphangiogenesis. a Ki-67 expression in the control rat kidney; b Ki-67 expression in the enalapril-treated rat kidney; c The enalapril-treated kidneys contained fewer Ki-67-positive proliferating cells compared with control rat kidneys (*P < 0.05 vs. controls); d VEGF-C expression in the control rat kidney; e VEGF-C expression in the enalapril-treated rat kidney; f The VEGF-C/β-actin protein expression was reduced significantly in the enalapril-treated rat kidney (*P < 0.05 vs. controls); g VEGFR-3 expression in the control rat kidney; h VEGFR-3 expression in the enalapril-treated rat kidney; i The VEGFR-3/β-actin protein expression was enhanced in the enalapril-treated rat kidney (*P < 0.05 vs. controls); j Podoplanin expression in the control rat kidney; and k Podoplanin expression in the enalapril-treated rat kidney (arrows, representative protein) (Black bar, control rats; white bar, enalapril-treated rats) (n = 8–12 for each group) ( × 400, Bar = 100 µm)
Intrarenal FGF-1, FGF-2, and FGFR-1 expression
We next examined the expression of FGF-1, FGF-2, and FGFR-1 (essential modulators of MET) in neonatal rat kidneys exposed to enalapril for the first 7 days.10 As depicted in Fig. 2, the expression of FGF-1, FGF-2, and FGFR-1 protein decreased after enalapril treatment on postnatal day 8. Immunoblots showed that the expression of FGF-1/β-actin, FGF-2/β-actin, and FGFR-1/β-actin decreased in the enalapril-treated group, compared with those in the control group (P < 0.05) (Fig. 2a, d and g). IHC analyses of normal control kidneys showed strong localization of FGF-1, FGF-2, and FGFR-1 in tubular epithelial cells and a few glomeruli (Fig. 2b, e and h). However, FGF-1 and FGFR-1 expression was rarely detected in the tubular cells of enalapril-treated kidneys (Fig. 2c and i). FGF-2 expression was also relatively weak in tubular epithelial cells in the enalapril-treated group (Fig. 2f), compared with the controls.

Intrarenal FGF-1 (a–c), FGF-2 (d–f), and FGFR-1 (g–i) expression. Enalapril-treated rat kidneys showed decreased intrarenal expression of FGF-1, FGF-2, and FGFR-1 compared with control rats (a, d, and g, immunoblots; black bar, control rats; white bar, enalapril-treated rats; *P < 0.05 vs. controls) (b, e, and h, control rats; c, f, and i, enalapril-treated rats; arrows, tubular expression; arrowheads, glomerular expression) (n = 8–12 for each group) ( × 400, Bar = 100 µm)
FSP-1 and α-SMA expression and interstitial collagen deposition
Due to the altered lymphangiogenesis accompanied by the reduced expression of MET modulators on day 8 in response to neonatal enalapril treatment, we investigated the localization of FSP-1 and α-SMA, well-known EMT markers.13 FSP-1 was not significantly localized in normal control kidneys as expected (Fig. 3a and c), whereas FSP-1 expression was prominent in a few glomerular and tubulointerstitial cells in the enalapril-treated neonatal kidneys (Fig. 3b and d). Compared with the controls, α-SMA expression was also highly detected within tubular epithelial cells of enalapril-treated kidneys (Fig. 3e and f), as shown earlier.18 Cortical areas of kidney sections stained with Sirius Red increased additionally in enalapril-treated rats, compared with control rats, indicating a higher renal interstitial collagen deposition in the enalapril-treated group (P < 0.001) (Fig. 3i). Enalapril-treated rats exhibited increased collagen deposition in periglomerular, tubulointerstitial, and perivascular areas of the kidney (Fig. 3g and h).

Intrarenal FSP-1 (a–d), α-SMA (e, f), and collagen (g–i) expression. Enalapril-treated rat kidneys (b, d) showed strong interstitial (arrows) and glomerular (arrowheads) FSP-1 expression compared with the control rat kidney (a, c). Intrarenal α-SMA expression were enhanced particularly in dilated tubular cells (arrows) in the enalapril-treated kidneys, compared with the controls (e, f). Cortical collagen deposition (arrows) was increased in the enalapril-treated rat kidneys compared with the control rat kidneys (*P < 0.05 vs. controls) (g–i). (a, c, e and g, control rats; b, d, f and h, enalapril-treated rats) (n = 8–12 for each group) (a, b × 100, Bar = 400 µm; c–f, × 400, Bar = 100 µm; g–h, Sirius Red stain, × 200, Bar = 200 µm)
Fibrinogen, ICAM-1, TLR-2, and TLR-4 expression
To determine whether additional fibrogenic molecules mediated renal fibrogenic signals following neonatal enalapril treatment, we further investigated the expression of fibrinogen (a novel candidate molecule in the development of renal fibrosis) and its three different cell surface receptors in renal fibroblasts: TLR-2, TLR-4, and ICAM-1.20 Enalapril-treated rats, however, exhibited no differences in intrarenal expression of fibrinogen, ICAM-1, TLR-2, and TLR-4, compared to control kidneys (Fig. 4a–d).

Immunoblots of renal fibrinogen (a), ICAM-1 (b), TLR-2 (c), and TLR-4 (d). No difference was observed in the intrarenal expression of fibrinogen, ICAM-1, TLR-2, and TLR-4, compared with the control kidneys (Black bar, control rats; white bar, enalapril-treated rats) (n = 8–12 for each group)
Discussion
The results of this study demonstrate that renal lymphangiogenesis was disrupted by neonatal enalapril treatment. Several markers for lymphatic vessels were downregulated in the developing rat kidneys following RAS blockade. Angiotensin converting enzyme (ACE) inhibition also reduced the protein expression of essential modulators of MET, namely FGF-1, FGF-2, and FGFR-1 in the developing newborn rat kidney. Intrarenal FSP-1, α-SMA, and interstitial collagen content quantified by Sirius Red staining was increased in the enalapril-treated rats. Our findings indicate that the generation of angiotensin II may be critical in normal renal lymphangiogenesis and homeostasis between tubular epithelial cells and interstitial fibroblasts. An altered epithelial–mesenchymal unit via RAS inhibition during kidney development may contribute to renal injury in this experimental model.
During nephrogenesis, lymphatic vessels migrate from the renal hilus deeply into the renal cortex along the kidney blood vessels.4 The extensive distribution of lymphatic capillaries in the renal cortex leads to interstitial fluid homeostasis.4 Although several endothelial markers of lymphatic vessels have been identified to date, there is currently no perfect and reliable marker of all disease states in different organs. In the developing mouse kidney, Tanabe et al.4 demonstrated the morphological characterization of lymphatic vessels in lymphangiogenesis via positive immunostaining for podoplanin, prox-1, and Ki-67. Nearly all lymphatic endothelial cells detected during lymphangiogenesis revealed Ki-67-positivity. In our study, Ki-67-positive proliferating cell numbers decreased and podoplanin-positive lymphatic vessels in the developing kidneys was reduced in response to neonatal enalapril treatment. VEGF-C, the best known lymphangiogenic growth factor, was also downregulated in the enalapril-treated rat kidneys. In contrast, the expression of its receptor, VEGFR-3, was upregulated by enalapril treatment. Because VEGF-C binding to VEGFR-3 induces lymphangiogenesis in vivo and stimulates proliferation, survival, and migration of lymphatic endothelial cells in vitro,19,21 altered VEGF-C/VEGFR-3 signaling may contribute to impaired lymphatic vessel development caused by ACE inhibition in the developing rat kidney.
The VEGFR system is critically involved in angiogenesis and lymphangiogenesis during vascular development and in neovascularization in adults.22 While VEGF-A and its receptors VEGFR-1/VEGFR-2 play key roles in both physiological and pathological angiogenesis, VEGF-C/VEGF-D and their receptor VEGFR-3 mostly act as critical regulators of lymphangiogenesis.23 Although VEGF-D also promotes lymphangiogenesis, its ability to induce lymphangiogenesis is less significant than that of VEGF-C.1 Clearly, the VEGF-C/VEGF-3 system has been recognized as the main signaling pathway to induce lymphangiogenesis.19 In this study, increased VEGFR-3 expression in the enalapril-treated kidneys may reflect a compensatory increase in receptor sensitivity in response to reduced activity of VEGF-C or be triggered by VEGF-D. While a full mechanistic explanation for the results is still lacking, these findings are consistent with our recent observations. Previously we showed that altered signaling between VEGF-A and VEGFR-1/VEGFR-2 and between angiopoietin and its receptor Tie-2 contributed to impaired angiogenesis during neonatal inhibition of ACE.17,24 The angiopoietin-Tie system regulates both cardiovascular and lymphatic development, inflammation, and pathological angiogenic responses.22 Given that lymphatic vessels facilitate the clearance of excess fluid and macromolecules from the interstitium and return them to the blood via lymph,25 disruption of lymphatic and blood vasculature network may contribute to the deterioration of tubulointerstitial compartment after neonatal RAS blockade in rats.
FGF ligands and FGFR are greatly expressed in the developing kidney. Renal organogenesis studies have identified FGF family of ligands and receptors as essential modulators of MET.10 Structural kidney and lower urinary tract diseases related to FGF or FGFR mutations have been reported in humans.26,27 Besides, FGF signaling plays a critical role in the development of both blood and lymphatic vasculature.12 FGFR-1 expressed in lymphatic endothelial cells is a critical receptor that mediates FGF-2–induced lymphangiogenesis. Interestingly, the VEGFR-3 signaling was a prerequisite for lymphatic tip cell formation in both FGF-2– and VEGF-C–induced lymphangiogenesis.11 With respect to renal pathology, FGF-2 exerts mostly proliferative effects on various renal cell types, although several aspects of FGF-2 in renal fibrogenesis remain poorly understood.11 While FGF-1 and FGFR-1 may be involved in inflammatory renal disease,28 the role of FGF-1 in kidney disease is also less defined. In our experiments, enalapril-treated neonatal kidneys showed reduced intrarenal levels of FGF-1, FGF-2, and FGFR-1. Decreased growth factor levels in neonatal ACE inhibition are not surprising since our serial data have continuously shown that ACE inhibition in the developing kidney reduces the renal expression of critical growth factors necessary for renal growth and development, such as TGF-β1 and epidermal growth factor.29,30,31 Reduced activities of FGF-1, FGF-2, and FGFR-1 may disrupt epithelial–mesenchymal crosstalk, lymphangiogenesis, and lymphatic and blood vasculature network in the developing kidney, which consistently contribute to renal growth impairment in conjunction with abnormal signaling of other essential growth factors/receptors following ACE inhibition during postnatal renal maturation. Multiple signaling pathways of kidney injury and repair may mediate defective homeostatic crosstalk in the epithelial–mesenchymal unit of developing kidney after the administration of enalapril.
Loss of the homeostatic crosstalk between tubular epithelial cells and fibroblasts (or mesenchymal cells) induces reorganization of the tubulointerstitial compartment. Proliferation and differentiation of fibroblasts into fully active myofibroblasts represents a key event in this altered microenvironment.13 Though EMT is crucial for appropriate re-epithelialization and ECM deposition in tissue repair, uncontrolled, and sustained myofibroblast activation leads to fibrosis.32 In tissue fibrosis, most epithelial cells express FSP-1, a representative fibroblast marker for identifying EMT, at an early stage.33,34 In this study we examined FSP-1 and α-SMA expression and interstitial collagen deposition. Our study shows that expression of FSP-1 and α-SMA was prominent in the tubulointerstitium and a few glomeruli in all kidneys treated with enalapril. Interstitial collagen deposition was also increased in the enalapril-treated kidneys. These findings are in accordance with our previous findings, which demonstrated that ACE inhibition increased renal EMT by upregulating α-SMA and vimentin and downregulating E-cadherin in the developing kidney.18 Many studies have documented EMT in tubular cells of human renal disease biopsies, and have correlated EMT features with the severity of the disease.35,36 The loss of renal function occurred with the proportional tubular cell expression of mesenchymal proteins, such as α-SMA, vimentin, and FSP-1, and these EMT markers predicted the prognosis of renal fibrosis.36,37 It can, therefore, be hypothesized that blockade of the RAS in animals with immature kidneys induces changes in epithelial–mesenchymal crosstalk, and these modifications contribute to the final fibrotic outcome.
In the present study, protein levels of fibrinogen and its different cell surface receptors on renal fibroblasts were also evaluated in newborn rat kidneys exposed to ACE inhibitor during postnatal nephrogenesis. Although fibrinogen, mediated by TLR-2, TLR-4, and ICAM-1, has been shown to mediate the development of renal fibrosis in the profibrotic animal model of unilateral ureteral obstruction,20 we were unable to detect increase in fibrinogen levels and its receptor protein synthesis after neonatal ACE inhibition. Additional experiments might be needed to test the fibrotic role of fibrinogen and its receptors during the time course of ACE inhibition in our experimental model.
Collectively, the current data suggest that impaired lymphangiogenesis and deteriorated MET/EMT signals may contribute to the abnormal renal development in the context of renal injury by neonatal ACE inhibition. Treatment of newborn rats with enalapril diminished renal cell proliferation, consistent with previous findings.38 In addition to its direct stimulation on renal cell growth, angiotensin II may regulate the renal expression of growth factors important for renal lymphangiogenesis and the balance between MET/EMT signals during postnatal nephrogenesis in rats. Altered interaction between tubular epithelial cells and mesenchymal cells, excessive accumulation of fluid and macromolecules in the interstitium due to impaired lymphatics, and rarefaction of peritubular capillaries may increase the risk of progressive renal injury in this experimental model. Although the role of pathologic lymphangiogenesis might vary with the renal damage,3 the creation of normal renal lymphatic circulation may play a crucial role in renal tubulointerstitial homeostasis during renal growth and development.
This study has a few limitations which need to be discussed. Deterioration of the renal lymphatic vasculature was not fully quantified. Stereological quantification of total length, volume, and surface area of renal lymphatic vessels may have contributed to greater statistical rigor in this study. The effects of impaired epithelial–mesenchymal crosstalk along with the disruption of kidney vasculature network on renal fibrosis need to be investigated long-term after neonatal RAS inhibition. Future studies also should examine the direct actions of angiotensin II on renal lymphangiogenesis and EMT/MET in kidney development as well as renal fibrosis and identify more elaborate mechanisms underlying kidney repair.
In summary, our data suggest that neonatal RAS inhibition during kidney development may induce alterations in tubulointerstitial compartment by impairing lymphangiogenesis and MET/EMT signals. A better understanding of the mechanisms regulating renal lymphatic functions and remodeling is necessary for renal health and in the management of renal diseases. Restoration of epithelial–mesenchymal homeostasis and architecture and/or modulation of molecular targets involved in the epithelial integrity and homeostasis may be an effective strategy to stop and/or even reverse progressive renal fibrosis.
References
Yazdani, S., Navis, G., Hillebrands, J. L., van Goor, H. & van den Born, J. Lymphangiogenesis in renal diseases: passive bystander or active participant? Expert. Rev. Mol. Med. 16, e15 (2014).
Ichise, T., Yoshida, N. & Ichise, H. FGF2-induced Ras–MAPK signalling maintains lymphatic endothelial cell identity by upregulating endothelial-cell-specific gene expression and suppressing TGFβ signalling through Smad2. J. Cell. Sci. 127, 845–857 (2014).
Hamar, P. & Kerjaschki, D. Blood capillary rarefaction and lymphatic capillary neoangiogenesis are key contributors to renal allograft fibrosis in an ACE inhibition rat model. Am. J. Physiol. Heart Circ. Physiol. 311, H981–H990 (2016).
Tanabe, M. et al. Development of lymphatic vasculature and morphological characterization in rat kidney. Clin. Exp. Nephrol. 16, 833–842 (2012).
Schulte-Merker, S., Sabine, A. & Petrova, T. V. Lymphatic vascular morphogenesis in development, physiology, and disease. J. Cell. Biol. 193, 607–618 (2011).
Karkkainen, M. J. et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 5, 74–80 (2004).
Schacht, V. et al. T1alpha/podoplanin deficiency disrupts normal lymphatic vasculature formation and causes lymphedema. EMBO J. 22, 3546–3556 (2003).
Cancilla, B., Davies, A., Cauchi, J. A., Risbridger, G. P. & Bertram, J. F. Fibroblast growth factor receptors and their ligands in the adult rat kidney. Kidney Int. 60, 147–155 (2001).
Powers, C. J., McLeskey, S. W. & Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 7, 165–197 (2000).
Chaffer, C. L., Thompson, E. W. & Williams, E. D. Mesenchymal to epithelial transition in development and disease. Cells Tissues Organs 185, 7–19 (2007).
Strutz, F. The role of FGF-2 in renal fibrogenesis. Front. Biosci. 1, 125–131 (2009).
Liang, G. et al. Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int. 93, 95–109 (2018).
Prunotto, M. et al. Epithelial–mesenchymal crosstalk alteration in kidney fibrosis. J. Pathol. 228, 131–147 (2012).
Trueb, B., Amann, R. & Gerber, S. D. Role of FGFRL1 and other FGF signaling proteins in early kidney development. Cell. Mol. Life Sci. 70, 2505–2518 (2013).
Guron, G. & Friberg, P. An intact renin-angiotensin system is a prerequisite for normal renal development. J. Hypertens. 18, 123–137 (2000).
Guron, G., Adams, M. A., Sundelin, B. & Friberg, P. Neonatal angiotensin converting enzyme inhibition in the rat induces persistent abnormalities in renal function and histology. Hypertension 29, 91–97 (1997).
Yoo, K. H., Yim, H. E., Bae, E. S. & Hong, Y. S. Capillary rarefaction and altered renal development: the imbalance between pro- and anti-angiogenic factors in response to angiotensin II inhibition in the developing rat kidney. J. Mol. Histol. 49, 219–228 (2018).
Yim, H. E., Yoo, K. H., Bae, I. S., Hong, Y. S. & Lee, J. W. Effect of angiotensin II inhibition on the epithelial to mesenchymal transition in developing rat kidney. Korean J. Pediatr. 52, 944–952 (2009).
Shibuya, M. & Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 312, 549–560 (2006).
Sörensen, I. et al. Fibrinogen, acting as a mitogen for tubulointerstitial fibroblasts, promotes renal fibrosis. Kidney Int. 80, 1035–1044 (2011).
Lohela, M., Bry, M., Tammela, T. & Alitalo, K. VEGFs and receptors involved in angiogenesis versus lymphangiogenesis. Curr. Opin. Cell Biol. 21, 154–165 (2009).
Eklund, L., Kangas, J. & Saharinen, P. Angiopoietin–tie signalling in the cardiovascular and lymphatic systems. Clin. Sci. 131, 87–103 (2017).
Shibuya, M. Vascular endothelial growth factor (vegf) and its receptor (vegfr) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2, 1097–1105 (2011).
Yim, H. E., Yoo, K. H., Bae, E. S., Hong, Y. S. & Lee, J. W. Impaired angiogenesis in the enalapril-treated neonatal rat kidney. Korean J. Pediatr. 59, 8–15 (2016).
Zhang, T. et al. Disturbance of lymph circulation develops renal fibrosis in rats with or without contralateral nephrectomy. Nephrology 13, 128–138 (2008).
Passos-Bueno, M. R. et al. Clinical spectrum of fibroblast growth factor receptor mutations. Hum. Mutat. 14, 115–125 (1999).
Walker, K. A., Sims-Lucas, S. & Bates, C. M. Fibroblast growth factor receptor signaling in kidney and lower urinary tract development. Pediatr. Nephrol. 31, 885–895 (2016).
Rossini, M. et al. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1), and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. 68, 2621–2628 (2005).
Choi, B. M. et al. Angiotensin-converting enzyme inhibition modulates mitogen-activated protein kinase family expressions in the neonatal rat kidney. Pediatr. Res. 57, 115–123 (2005).
Kang, N. S. et al. ACE inhibition modulates transforming growth factor-beta receptors in the young rat. Pediatr. Nephrol. 18, 865–871 (2003).
Yoo, K. H., Wolstenholme, J. T. & Chevalier, R. L. Angiotensin-converting enzyme inhibition decreases growth factor expression in the neonatal rat kidney. Pediatr. Res. 42, 588–592 (1997).
Stone, R. C. et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 365, 495–506 (2016).
Iwano, M. et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 110, 341–350 (2002).
Zeisberg, E. M. et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961 (2007).
Zheng, M. et al. Urinary mRNA markers of epithelial–mesenchymal transition correlate with progression of diabetic nephropathy. Clin. Endocrinol. 76, 657–664 (2012).
Hazzan, M. et al. Epithelial-to-mesenchymal transition predicts cyclosporine nephrotoxicity in renal transplant recipients. J. Am. Soc. Nephrol. 22, 1375–1381 (2011).
Xu-Dubois, Y. C. et al. Epithelial phenotypic changes are associated with a tubular active fibrogenic process in human renal grafts. Hum. Pathol. 44, 1251–1261 (2013).
McCausland, J. E., Ryan, G. B. & Alcorn, D. Angiotensin converting enzyme inhibition in the postnatal rat results in decreased cell proliferation in the renal outer medulla. Clin. Exp. Pharmacol. Physiol. 23, 552–554 (1996).
Acknowledgements
This study was supported by a Korea University Grant.
Author information
Authors and Affiliations
Contributions
Substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; K.H.Y., H.E.Y., and E.S.B. Drafting the article or revising it critically for important intellectual content: K.H.Y. and H.E.Y. Final approval of the version to be published: K.H.Y., H.E.Y., E.S.B. and Y.S.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yoo, K.H., Yim, H.E., Bae, E.S. et al. Angiotensin inhibition in the developing kidney; tubulointerstitial effect. Pediatr Res 85, 724–730 (2019). https://doi.org/10.1038/s41390-019-0288-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0288-9
