
Abstract
The glutamatergic modulator ketamine has striking and rapid antidepressant effects in major depressive disorder (MDD), but its mechanism of action remains unknown. Proton magnetic resonance spectroscopy (1H-MRS) is the only non-invasive method able to directly measure glutamate levels in vivo; in particular, glutamate and glutamine metabolite concentrations are separable by 1H-MRS at 7T. This double-blind, placebo-controlled, crossover study that included 1H-MRS scans at baseline and at 24 h post ketamine and post-placebo infusions sought to determine glutamate levels in the pregenual anterior cingulate (pgACC) of 20 medication-free MDD subjects and 17 healthy volunteers (HVs) 24 h post ketamine administration, and to evaluate any other measured metabolite changes, correlates, or predictors of antidepressant response. Metabolite levels were compared at three scan times (baseline, post-ketamine, and post-placebo) in HVs and MDD subjects at 7T using a 1H-MRS sequence specifically optimized for glutamate. No significant between-group differences in 1H-MRS-measured metabolites were observed at baseline. Antidepressant response was not predicted by baseline glutamate levels. Our results suggest that any infusion-induced increases in glutamate at the 24-h post ketamine time point were below the sensitivity of the current technique; that these increases may occur in different brain regions than the pgACC; or that subgroups of MDD subjects may exist that have a differential glutamate response to ketamine.
Similar content being viewed by others

Introduction
Major depressive disorder (MDD) is a devastating illness with a poorly understood biological etiology. A growing body of pre-clinical and clinical evidence implicates the glutamatergic neurotransmitter system in the pathophysiology and treatment of MDD [1]. In addition, recent data have consistently suggested that the glutamatergic modulator ketamine has striking and rapid antidepressant effects [2,3,4,5], underscoring the role of glutamate in the treatment and pathophysiology of MDD [6, 7].
Currently, the only technique capable of directly and non-invasively investigating underlying glutamatergic pathophysiology in vivo is magnetic resonance spectroscopy (MRS). Several previous proton MRS (1H-MRS) investigations in MDD subjects detected lower levels of glutamatergic metabolites (glutamate and glutamine) compared with healthy volunteers (HVs) [8, 9]; however, results have occasionally been inconsistent [10]. Recent 1H-MRS investigations noted increased glutamatergic metabolite levels in HVs [11, 12] and increased Glx (a composite measure encompassing both glutmate and glutamine) levels in MDD subjects [13] following acute ketamine administration; the ratio of glutamine to glutamate was also found to be increased up to a day after acute ketamine administration in HVs [14]. Nevertheless, other investigations found no significant changes in glutamate measurements 1 h post ketamine infusion in HVs [15], or three and 48 h later in MDD subjects [16]; variations in voxel location, medication status, timing of the scan, imaging parameters, and sample size may explain these discrepant findings (Supplementary Table S1 summarizes parameters for these papers). In addition, one study found that treatment response to ketamine was predicted by baseline levels of glutamatergic metabolites [17]. Taken together, these findings suggest that understanding the function of the glutamatergic system during major depressive episodes may be key to effectively treating MDD.
Most 1H-MRS studies to date conducted in MDD subjects have used 1.5, 3, or 4T magnetic resonance imaging (MRI) scanners, which may not adequately distinguish glutamatergic metabolites. We recently developed a reliable [18] and effective [19] 1H-MRS sequence capable of delineating glutamate and glutamine from each other and other neural metabolites at 7T. Here we compared glutamate and glutamine metabolites in the pregenual anterior cingulate cortex (pgACC) in treatment-resistant, medication-free MDD subjects, as well as in HVs using our 1H-MRS sequence at 7T; glutamatergic metabolism is thought to be altered in this area of the cortex in MDD subjects [20]. After baseline imaging assessments, MDD subjects and HVs received infusions of both ketamine and placebo (2 weeks apart) and were scanned approximately 24-h post infusion (following both ketamine and placebo administration) using the same imaging parameters.
Given the recent positive findings in the 1H-MRS depression literature [8, 9, 21, 22], we expected to observe decreased glutamate levels in MDD subjects relative to HVs. We further anticipated that ketamine would increase levels of glutamatergic metabolites in the pgACC 24 h post infusion to a degree correlated with drug response in MDD subjects and that would not affect other metabolites.
This study is part of a larger study designed to identify clinical and neurobiological correlates of ketamine treatment in unmedicated inpatients with treatment-resistant MDD. Here we assessed the effects of ketamine on mood and brain glutamate changes measured at baseline and at 24 h post ketamine/post placebo infusion.
Materials and methods
Participants
All subjects were recruited as part of a larger study (NCT#00088699, NIH Protocol #04-M-0222, sub-study 4). Twenty MDD subjects and 17 HVs who had 1H-MRS scans were included in this analysis. All participants were between 18 and 65 years old (see Table 1). Each subject provided written informed consent as approved by the National Institutes of Health (NIH) Combined Central Nervous System Institutional Review Board.
All subjects were physically healthy, as determined by medical history, physical examination, blood labs, urinalysis, and toxicology, and free of any serious medical conditions, comorbid substance abuse (within the preceding 3 months), or lifetime dependence (excluding caffeine and nicotine). HV and MDD subjects were evaluated and diagnosed via the structured clinical interview for DSM-IV Axis I disorders [23, 24] and an unstructured interview with a board-certified psychiatrist. HVs were excluded if they had any first-degree relatives diagnosed with a major (Axis I) psychiatric disorder. MDD subjects were admitted to the 7SE inpatient unit at the National Institutes of Health Clinical Research Center for the duration of the study. HVs were outpatients but received all study-related interventions on the unit. Three MDD subjects and one HV were smokers at the time of the study.
All MDD subjects were currently experiencing a major depressive episode lasting at least 4 weeks and had been medication-free for at least 2 weeks prior to study randomization. A Montgomery-Åsberg Depression Rating Scale (MADRS) [25] score ≥20 at the time of screening and also prior to each infusion was an inclusion criterion for MDD subjects. In addition, MDD subjects were refractory to pharmacological treatment; treatment-resistant was defined as not having responded to at least one adequate antidepressant dose/duration trial (as defined in ref. [26]).
Design
Figure 1a illustrates the randomized, double-blind, placebo-controlled, crossover design of this study. After a medication taper (if necessary) and a minimum 2-week drug-free period, participants received one intravenous infusion of a sub-anesthetic dose of ketamine hydrochloride (0.5 mg/kg) and one infusion of placebo (0.9% saline solution), with 2 weeks between infusions. Infusion order was randomized across participants. The infusions were administered by an advanced cardiac life support-licensed practitioner over 40 min on the inpatient unit; infusion solutions appeared identical to ensure blinding. No psychotherapy or other treatment was permitted during the entire trial period.

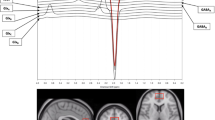
a Schematic of the double-blind, placebo-controlled, crossover study illustrating the timing of the medication taper, ketamine and placebo infusions (triangle), and magnetic resonance spectroscopy (MRS) imaging (circle). b (i) Example voxel location (yellow box) shown overlaid on an anatomical image slice in the saggital (left), axial (middle), and coronal directions. (ii) Centroid locations of all MRS voxels overlaid on a glass brain. (iii) Example segmentation into gray matter (GM), white matter (WM), and cerebrospinal fluid (CSF) with overlaid voxel location in red. (c) Example voxel spectrum labeled with modeled metabolites
Psychometric scales
Mood ratings were obtained using the following clinician-administered rating scales: the MADRS, the Hamilton Depression Rating Scale (HAM-D) [27], and the Snaith-Hamilton Pleasure Scale (SHAPS) [28]. Ratings were obtained 60 min prior to both ketamine and placebo infusions and on the days of the 1H-MRS scans.
MRI
MRI scans were acquired using a Siemens 7T Magnetom MRI scanner (Erlangen, Germany) with a 32-channel head coil. Scans took place at baseline (about 3 days before the first infusion) and 24 h after both ketamine and placebo infusions. Standard 1 mm3 isotropic resolution magnetization-prepared rapid gradient echo sequence (MPRAGE) images (repetition time (TR): 3 s, echo time (TE): 3.9 ms) were acquired on each of the scanning days and used to create anatomical images that were used to plan the location of the 1H-MRS voxel.
1H-MRS
The precise parameters of our imaging protocol have been detailed elsewhere [18, 19]. Briefly, a 2 cm isotropic voxel was acquired from the pgACC (Fig. 1b) using a TE-optimized point resolved spectroscopy (PRESS) sequence with TE1 = 69 ms, TE2 = 37 ms with an inserted 90° J-suppression radio-frequency pulse to minimize the N-acetyl-aspartate (NAA) multiplet at 2.5 p.p.m. [19] (additional sequence parameters: TR: 2.5 s, spectral width: 4000 Hz, number of data points: 2048, number of transients: 128 with water suppression using eight radio-frequency pulses of 350 Hz bandwidth). The time-averaged 32-channel free induction decay (FID) signals were merged into a combined single-channel metabolite FID using a generalized least squares method [29]. Metabolite levels were then estimated from the spectrum (Fourier transform of the combined FID) using a custom-written linear combination fitting program [19]. Basis sets included glutamate, glutamine, glutathione, γ-aminobutyric acid (GABA), N-acetyl aspartate (NAA), N-acetylaspartylglutamate (NAAG), choline, and creatine. For statistical analyses, NAA was summed with NAAG (henceforth known as total NAA (tNAA)) because the sequence used here is not optimal for reliably detecting NAAG. Similarly, because the sequence is not ideal for measuring GABA, GABA was modeled but not reported.
Metabolite values are presented relative to creatine because creatine levels are considered relatively constant across individuals and because no known differences in creatine levels have been observed between MDD subjects and HVs [30].
A water proton peak-line broadening criterion of 16 Hz was selected, and only spectra with less than this value were included in the analyses to ensure high-quality data. A representative 1H-MRS spectrum obtained using our sequence is presented in Fig. 1c.
Tissue segmentation and voxel localization
The anatomical image was segmented into gray matter (GM), white matter (WM), and cerebrospinal fluid (CSF) components using the FSL tool FAST [31]. The MRS voxel region of interest (ROI) was generated by creating a 20 mm cube mask centered at the voxel coordinates in subject space (3dcalc). The tissue composition of the voxel was then calculated using the segmented anatomical (3dROIstats).
In order to visualize all voxel centroids in a single-reference space, each scan’s voxel coordinates were first obtained from the MRS raw data file. The high-resolution anatomical image from which the MRS voxel was prescribed was then warped to standard Montreal Neurological Institute (MNI)-152 space (@auto_tlrc in AFNI [32]). The resulting linear transformation matrix was then applied to the center coordinate of the voxel to obtain the coordinate in standard MNI space.
Statistical analyses
A linear mixed effects model (LME) was performed in R (R [33]) (using lme from the package nlme with restricted log-likelihood (REML) model fitting) at the metabolite level in order to evaluate the group and scan effects with subject as a random variable. Further models, including age, gray matter (GM), and MADRS as covariates were also explored. Post hoc contrasts evaluating pairwise metabolite differences between scans (ketamine-baseline, placebo-baseline, ketamine-placebo) were carried out using testInteractions from the package phia, using a χ2-test for this type of model [34], with multiple comparison correction ‘holm’ (i.e., Holm-Bonferroni correction). A separate model was used to evaluate behavioral measures. Pearson product moment correlation coefficients were used to assess the relationship between behavioral measures and metabolite levels. Significance was set at p < 0.05.
Results
Data descriptors and demographics
Table 2 shows the number of scans completed per session along with the average water line width and signal to noise ratio (SNR). Two HV placebo scans were excluded for having line widths greater than 16 Hz. The HV and MDD groups did not differ in age (F(1,35) = 0.04, p = 0.83) or water line width (F(1,35) = 0.97, p = 0.33). No interaction effect of scan by group for water line width was observed.
Behavioral response to ketamine
The mean change in mood rating scores for both groups at each scan time point is summarized in Table 1. Significant differences in behavioral measures were noted in the MDD group between the post-ketamine infusion and baseline scans (MADRS:χ2 = 23.7, p < 0.001, HAM-D:χ2 = 25.7, p < 0.001, SHAPS:χ2 = 5.1, p < 0.04), as well as between post-ketamine and placebo scans (MADRS:χ2 = 12.5, p < 0.001, HAM-D:χ2 = 4.8, p = 0.02, SHAPS:χ2 = 9.1, p < 0.001). No significant differences in mood were observed for these rating scales in the HV dataset. Results from the full dataset demonstrated a slight but significant increase in depressive symptoms in HVs; for details, please see ref. [35].
1H-MRS
Figure 2a illustrates the individual and HV and MDD group mean metabolite values for glutamate across scans (all other modeled metabolites are shown in Supplementary Figure S1 for completeness). Figure 2b displays the glutamate values across scans for each MDD subject; these are linked by lines to better illustrate individual variations in reponse to ketamine. Subjects were color coded depending on whether they exhibited increased or decreased glutamate post-ketamine from baseline.

a Group mean glutamate (Glu) concentrations referenced to creatine for the healthy volunteer (HV, red) and major depressive disorder (MDD, blue) groups for each scan. Each filled circle represents the value for a subject. The open circle is the group mean, and the error bars are the standard error. b Glutamate values for each MDD subject are shown joined with a line across scans. Solid dark blue lines are subjects where the glutamate value was larger at baseline than after ketamine administration and the light dotted blue line are subjects whose glutamate levels increased after ketamine (only subjects with completed baseline scans are shown)
The results below were drawn from the simplest model with no covariates, given that all exploratory models returned similar group comparison statistics. No significant effects were noted for mean glutamate levels at baseline (HV = 1.25 ± 0.02; MDD = 1.29 ± 0.02; HV-MDD = −0.04, p = 0.48, uncorrected), post-ketamine administration (HV = 1.28 ± 0.03; MDD = 1.36 ± 0.03; HV-MDD = −0.08, p = 0.2, uncorrected), post-placebo administration (HV = 1.27 ± 0.03; MDD = 1.32 ± 0.03; HV-MDD = −0.05, p = 0.4, uncorrected), or any other modeled metabolites (full listing appears in Table 3). In addition, no significant relationship was observed in the MDD group between baseline glutamate levels and pre-infusion MADRS score (r(16) = −0.15, p = 0.53).
Pairwise scan differences (baseline-ketamine, baseline-placebo, ketamine-placebo) were compared within each group in order to identify changes resulting from ketamine administration (see Table 4 for details). Although, the MDD group showed a nominal glutamate increase in the post-ketamine scans compared to both baseline (value = 0.07, p = 0.1) and placebo scans (value = 0.04, p = 0.1), these were not significant changes. Similarly, tNAA levels were somewhat increased following ketamine administration compared to both baseline (value = 0.04, p = 0.05) and post-placebo values (value = 0.04, p = 0.05).
Figure 2b shows that of the five subjects exhibiting an unexpected decrease in glutamate levels post-ketamine compared to baseline (dark blue lines), four also had the highest baseline glutamate levels. The subjects exhibiting increased glutamate levels post-ketamine administration compared to baseline (light blue dashed lines) generally had lower baseline glutamate levels. This preliminary and exploratory observation may potentially indicate functional subgroups of MDD subjects.
Discussion
This 7T 1H-MRS study investigated glutamate and glutamine levels in the pgACC in both unmedicated, treatment-resistant MDD subjects and HVs at baseline and at 24 h after a single infusion of intravenous ketamine and placebo. Contrary to our expectations, we found no significant differences in metabolites, either between groups or between scan sessions, although MDD subjects did have a trend toward increased glutamate post-ketamine infusion. In addition, no correlations were observed between glutamate levels and mood, and baseline glutamate levels were not associated with subsequent response to ketamine.
Previous studies have reported decreased glutamate levels in subjects with severe MDD (as defined using HAM-D scores) compared to HVs in the anterior cingulate cortex (ACC) [8, 36], but these results may vary by depression subtype [36] and in response to the exact location of the 1H-MRS voxel. Horn et al.[36] separated MDD subjects into two groups using a HAM-D score of 15 and found that only the more severely depressed subjects showed reduced glutamate levels. Given that our MDD group had an average HAM-D score of 21.8 at baseline, it should be comparable to the more severely depressed HAM-D >15 group of Horn et al.[36]; however, in contrast to the results of Horn et al.[36] and to our own expectations, we found a trend toward increased glutamate levels in MDD subjects compared to HVs. Several differences between the MDD subjects in our study and that of Horn et al.[36] may have contributed to these results, including the unmedicated and psychotherapy-free status of our MDD group.
Unlike most previously published studies, the sequence used here at 7T permits resolution of the glutamate peak as distinct from glutamine. Furthermore, the J-edited PRESS sequence was optimized for glutamate detection [19], such that our measurements should be relatively free of confounding effects from surrounding metabolites, with additional benefits from increased signal-to-noise at this higher field strength. Despite these strengths, we found no significant differences in any measured metabolite levels between MDD subjects and HVs at baseline or between post-ketamine and baseline for either group; we observed only a trend-level increase in glutamate levels post-ketamine relative to both baseline and the post-placebo scan. Our results agree with a study by Valentine et al.[16] that found no changes in metabolite levels either 3 h or 2 days post ketamine infusion in the occipital cortex of 10 subjects with MDD. Taylor et al.[15] measured ACC metabolite levels in HVs at 3T at baseline, as well as during and immediately after a 40-min ketamine infusion and similarly found no changes in metabolite levels. Interestingly, an estimate for the Cohen’s d effect size of the difference in glutamate levels post-ketamine vs post-placebo in the present study was 0.15, which is similar to that obtained by Valentine et al.[16] in the occipital cortex. This study also identified a trend-level increase in tNAA, which has not previously been noted in response to ketamine. Given that NAA is frequently taken as a marker of neuronal integrity [37], this finding may be worthy of future study. It should also be pointed out that we were measuring steady-state glutamate levels averaged over ~6 min from a relatively large (2 cm3) volume of cortex, which reduces our ability to specifically localize the source of the glutamate changes. Approaches using 13C MRS to directly measure the glutamate-glutamine cycle [10], as well as increasing the sensitivity of the MRS technique, may help elucidate these details.
A noteworthy observation about the glutamate changes seen in our group of MDD subjects is the apparent difference in response to ketamine between the group of subjects with higher and lower baseline glutamate values. This suggests the existence of distinct MDD subgroups: one that experiences the expected increase in glutamate levels after ketamine administration and one that does not. The finding raises a potential avenue for future investigation into using baseline glutamate values as a way to identify MDD subgroups.
Interestingly, Li et al.[14] found an increased glutamine/glutamate ratio measured at 7T in the pgACC in HVs 24 h post ketamine administration, a finding not reproduced here. Their study used a stimulated echo acquisition mode (STEAM) sequence with a short TE where the value measured for the glutamine peak may have been more affected by the presence of macromolecules than our measurement due to the long TE used in our sequence. It should be noted that other brain regions may exhibit larger glutamate changes than the pgACC. Futhermore, some, but not all, studies have found reduced GM volumes in the pgACC and subgenual ACC (sgACC) in MDD subjects [38], which may also account for some variance; however, including GM as a covariate in the LME model did not result in a significant main effect of scan session (data not shown).
Several possibilities may account for our observed lack of a measurable difference at 24 h post ketamine. First, the glutamate difference induced by ketamine at this time point may be lower than the 8% sensitivity of our MRS sequence [18]; second, the ketamine-induced proposed glutamate burst may happen before 24 h, may be acute, and may be responsible for ketamine’s acute rather than sustained antidepressant effects; and third, glutamate differences might not occur in the pgACC. One study did find combined glutamate and glutamine (Glx) changes within the first 30 min post ketamine administration in the pgACC [13], which supports the idea that the glutamate burst happens quite early post-ketamine infusion. While the sgACC is more frequently implicated in the pathophysiology of MDD than the pgACC, spectra with acceptable linewidths are generally not possible in this brain region due to its proximity to the sinuses. Other studies have found ketamine-induced glutamatergic differences in the hippocampus [39] and thalamus [12]. Thus, it is possible that future experiments should focus on earlier time points or other brain regions.
This study is associated with several strengths. First, we studied both unmedicated treatment-resistant MDD subjects as well as HVs. Second, we acquired 1H-MRS measurements at the high field strength of 7T, where glutamate and glutamine are separable, in both groups. Third, both subject groups received the same scans and administration of ketamine and placebo infusions, which enabled direct comparison of their responses. However, and despite the intriguing nature of the results, the present study is also associated with several limitations. First, the cohort size was modest, which limited our ability to draw a definitive conclusion from our results. Second, a small amount of variance was incurred from manually positioning the voxel; however, the measured voxel placement difference was much smaller than the voxel size itself. Third, we did not monitor motion—which might have decreased the measured metabolite values—during these long acquisitions; nevertheless, the variance within the MDD and HV data was comparable, suggesting similar measurement conditions between the groups. Finally, we chose to reference our metabolite values to creatine, which may not be ideal given the possibility of inter-individual, regional, or disease variance. However, motion will have a larger effect on metabolite/water ratio than metabolite/creatine ratio as the water signal is obtained separately and, as such, may not constitute a better reference. Future studies may consider measuring the stability of creatine values in the pgACC in MDD.
Conclusion
In order to assess underlying glutamate changes associated with ketamine administration in both unmedicated, treatment-resistant MDD subjects and HVs, we acquired 1H-MRS measurements at 7T in both groups. We found that 7T 1H-MRS pgACC glutamate differences between HVs and MDD subjects in this study were less than the measurement sensitivity of the pulse sequence (~8% change). The data suggest that larger effects may occur earlier than 24 h or in a different brain region, or that subgroups of MDD subjects may exist that have a differential response to ketamine. Our findings may help guide future investigations of the neurochemical changes post ketamine administration in individuals with MDD.
References
Sanacora G, Treccani G, Popoli M. Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology. 2012;62:63–77.
Dutta A, McKie S, Deakin JF. Ketamine and other potential glutamate antidepressants. Psychiatry Res. 2015;225:1–13.
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4.
Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–802.
Jaso BA, Niciu MJ, Iadarola ND, Lally N, Richards EM, Park M, et al. Therapeutic modulation of glutamate receptors in major depressive disorder. Curr Neuropharmacol. 2017;15:57–70.
Murrough JW, Abdallah CG, Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov. 2017;16:472–86.
Luykx JJJ, Laban KGG, van den Heuvel MPP, Boks MPMP, Mandl RCWC, Kahn RSS, et al. Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of (1)H-MRS findings. Neurosci Biobehav Rev. 2012;36:198–205.
Yuksel C, Ongur D, Yüksel C, Öngür D. Magnetic resonance spectroscopy studies of glutamate-related abnormalities in mood disorders. Biol Psychiatry. 2010;68:785–94.
Abdallah CG, Jiang L, Feyter HM De, Fasula M, Krystal JH, Rothman DL, et al. Glutamate metabolism in major depressive disorder. Am J Psychiatry. 2014;171:1320–7.
Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–6.
Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–5.
Milak MS, Proper CJ, Mulhern ST, Parter AL, Kegeles LS, Ogden RT, et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol Psychiatry. 2015;21:320–7.
Li M, Demenescu LR, Colic L, Metzger CD, Heinze H-J, Steiner J, et al. Temporal dynamics of antidepressant ketamine effects on glutamine cycling follow regional fingerprints of AMPA and NMDA receptor densities. Neuropsychopharmacology. 2017;42:1201–9.
Taylor MJ, Tiangga ER, Mhuircheartaigh RN, Cowen PJ. Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. J Psychopharmacol. 2012;26:733–7.
Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Res. 2011;191:122–7.
Salvadore G, Veen JW, van der, Zhang Y, Marenco S, Machado-Vieira R, Baumann J, et al. An investigation of amino-acid neurotransmitters as potential predictors of clinical improvement to ketamine in depression. Int J Neuropsychopharmacol. 2012;15:1063–72.
Lally N, An L, Banerjee D, Niciu MJ, Luckenbaugh DA, Richards EM, et al. Reliability of 7T 1H-MRS measured human prefrontal cortex glutamate, glutamine, and glutathione signals using an adapted echo time optimized PRESS sequence: A between- and within-sessions investigation. J Magn Reson Imaging. 2016;43:88–98.
An L, Li S, Murdoch JB, Araneta MF, Johnson C, Shen J. Detection of glutamate, glutamine, and glutathione by radiofrequency suppression and echo time optimization at 7 tesla. Magn Reson Med. 2015;73:451–8.
Walter M, Henning A, Grimm S, Schulte RF, Beck J, Dydak U, et al. The relationship between aberrant neuronal activation in the pregenual anterior cingulate, altered glutamatergic metabolism, and anhedonia in major depression. Arch Gen Psychiatry. 2009;66:478–86.
Li M, Walter M. The acute and chronic effects of ketamine as revealed by noninvasive brain imaging. Neuropathol Drug Addict Subst Misuse. 2016; 689–702. https://doi.org/10.1016/B978-0-12-800212-4.00064-9.
Taylor M. MRS of Psychiatric Disorders. 2013 Chapter 3.6. https://doi.org/10.1016/B978-0-12-401688-0.00016-1.
First, Michael B., Spitzer, Robert L, Gibbon Miriam, and Williams, Janet B.W.: Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition. (SCID-I/P) New York: Biometrics Research, New York State Psychiatric Institute, November 2002.
First, Michael B., Spitzer, Robert L, Gibbon Miriam, and Williams, Janet B.W.: Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Non-patient Edition. (SCID-I/NP) New York: Biometrics Research, New York State Psychiatric Institute, November 2002.
Montgomery SA, Åsberg M, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9.
Sackeim HA. The definition and meaning of treatment-resistant depression. J Clin Psychiatry. 2001;62:10–17.
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62.
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry. 1995;167:99–103.
An L, Willem van der Veen J, Li S, Thomasson DM, Shen J. Combination of multichannel single-voxel MRS signals using generalized least squares. J Magn Reson Imaging. 2013;37:1445–50.
Buonocore MH, Maddock RJ. Magnetic resonance spectroscopy of the brain: a review of physical principles and technical methods. Rev Neurosci. 2015;26:609–32.
Zhang Y, Brady M, Smith S. Segmentation of brain MR images through a hidden Markov random field model and the expectation-maximization algorithm. IEEE Trans Med Imaging. 2001;20:45–57.
Cox R. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162–73.
R Core Team. R: A Language and Environment for Statistical Computing. (2013). Available at http://www.r-project.org/.
Fox J, Weisberg S. An {R} Companion to Applied Regression. Thousand OAks, CA: SAGE Publications; 2011.
Nugent A, Ballard E, Gould T, Park LT, Moaddel R, Brutsche N, et al. Ketamine has distinct electrophysiological and behavioural effects in depressed and healthy subjects. Mol Psychiatry. 2018. https://doi.org/10.1038/s41380-018-0028-2. [Epub ahead of print] https://www.ncbi.nlm.nih.gov/pubmed/29487402
Horn DI, Yu C, Steiner J, Buchmann J, Kaufmann J, Osoba A, et al. Glutamatergic and resting-state functional connectivity correlates of severity in major depression - the role of pregenual anterior cingulate cortex and anterior insula. Front Syst Neurosci. 2010;4:33.
Baslow MH. N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochem Res. 2003;28:941–53.
Drevets WC, Savitz J, Trimble M. The subgenual anterior cingulate cortex in mood disorders. CNS Spectr. 2008;13:663–81.
Kraguljac NV, Frölich MA, Tran S, White DM, Nichols N, Barton-McArdle A, et al. Ketamine modulates hippocampal neurochemistry and functional connectivity: a combined magnetic resonance spectroscopy and resting-state fMRI study in healthy volunteers. Mol Psychiatry. 2017;22:562–9.
Acknowledgements
The authors thank the 7SE research unit and staff for their support. Ioline Henter (NIMH) provided invaluable editorial assistance
Funding
Funding for this work was supported by the Intramural Research Program at the National Institute of Mental Health, National Institutes of Health (IRP-NIMH-NIH; ZIA MH002857), by a NARSAD Independent Investigator Award to Dr. Zarate, and by a Brain and Behavior Mood Disorders Research Award to Dr. Zarate. Dr. Zarate is listed as a co-inventor on a patent for the use of ketamine and its metabolites in major depression and suicidal ideation. Dr. Zarate is listed as a co-inventor on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydro and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain. Dr. Zarate is listed as co-inventor on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post-traumatic stress disorders; he has assigned his patent rights to the US government but will share a percentage of any royalties that may be received by the government.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jennifer W. Evans, Níall Lally
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Evans, J.W., Lally, N., An, L. et al. 7T 1H-MRS in major depressive disorder: a Ketamine Treatment Study. Neuropsychopharmacol 43, 1908–1914 (2018). https://doi.org/10.1038/s41386-018-0057-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0057-1
This article is cited by
-
Ketamine and rapid antidepressant action: new treatments and novel synaptic signaling mechanisms
Neuropsychopharmacology (2024)
-
Elevating the field for applying neuroimaging to individual patients in psychiatry
Translational Psychiatry (2024)
-
Ketamine and Zinc: Treatment of Anorexia Nervosa Via Dual NMDA Receptor Modulation
CNS Drugs (2023)
-
Acute effects of ketamine on the pregenual anterior cingulate: linking spontaneous activation, functional connectivity, and glutamate metabolism
European Archives of Psychiatry and Clinical Neuroscience (2022)
-
The potential of 1H-MRS in CNS drug development
Psychopharmacology (2021)
