
Abstract
The classification of adenohypophysial neoplasms as “pituitary neuroendocrine tumors” (PitNETs) was proposed in 2017 to reflect their characteristics as epithelial neuroendocrine neoplasms with a spectrum of clinical behaviors ranging from small indolent lesions to large, locally invasive, unresectable tumors. Tumor growth and hormone hypersecretion cause significant morbidity and mortality in a subset of patients. The proposal was endorsed by a WHO working group that sought to provide a unified approach to neuroendocrine neoplasia in all body sites. We review the features that are characteristic of neuroendocrine cells, the epidemiology and prognosis of these tumors, as well as further refinements in terms used for other pituitary tumors to ensure consistency with the WHO framework. The intense study of PitNETs has provided information about the importance of cellular differentiation in tumor prognosis as a model for neuroendocrine tumors in different locations.
Similar content being viewed by others

Introduction
Pituitary tumors include neoplasms that arise in and around the sella turcica [1, 2]. The majority arise from the hormone-secreting adenohypophysial cells that are members of the family of neuroendocrine epithelial cells. Other tumors include those arising from pituicytes (the modified glia of the posterior lobe), tumors that arise from the meninges, stromal elements, and adjacent hypothalamus, as well as craniopharyngiomas and many others [3].
Tumors of adenohypophysial cells exhibit a wide spectrum of hormonal and proliferative characteristics; small tumors may be incidental findings [4] or may be the cause of significant morbidity and even mortality, as in the case of untreated Cushing disease [5]. A large number of tumors are prolactinomas (functional lactotroph tumors) that respond to medical therapy with normalization of hormone levels and tumor shrinkage in the majority of cases. However, a significant proportion, estimated to be more than half of all patients with clinically diagnosed anterior pituitary tumors [6], require surgical intervention, some with curative intent, others for debulking and avoidance of compressive effects. Up to one quarter of patients who undergo surgery have residual and persistent disease that may entail complex management for hormone regulation as well as additional surgical intervention, radiotherapy, and chemotherapeutic attempts to restrain tumor growth.
In this review, we summarize the features of adenohypophysial hormone-producing cells that represent neuroendocrine cells, we discuss the terminologies that are relevant for the classification of the neoplasms that arise from these cells, and we document the epidemiology of these lesions. The large body of work reviewing prognostic features of the various tumor types is summarized and the importance of tumor subtyping is discussed as a model for other neuroendocrine tumors (NETs).
Hormone-secreting pituitary adenohypophysial cells are neuroendocrine cells
Endocrine cells that produce hormones are found throughout the body, both in endocrine glands and dispersed among other cells of non-endocrine organs. They fall into three categories based on cell differentiation and the biochemical features of the hormone(s) they produce [7]:
-
(1)
Steroid hormone-producing cells are found mainly in the adrenal cortex and gonads. They are thought to be of mesodermal embryonic origin and their biochemistry involves the uptake of cholesterol for conversion by complex enzymatic processes into glucocorticoid, mineralocorticoid, and sex steroid hormones. They are characterized structurally by an abundance of smooth endoplasmic reticulum and fat globules.
-
(2)
Thyroid follicular cells, of endodermal origin, are epithelial cells that synthesize thyroglobulin, store it in sequestered follicles, and resorb it for iodination and cleavage into thyroid hormones. They have extremely well-developed intercellular junctions, rough endoplasmic reticulum, and complex microvilli at the apical lumen that are critical for resorption of thyroglobulin.
-
(3)
The largest family of endocrine cells have neuroendocrine differentiation and are known as neuroendocrine cells (Fig. 1). These cells all are involved in the production of peptide hormones that are produced in the rough endoplasmic reticulum, processed in the Golgi complex and packaged into membrane-bound neurosecretory granules where they are stored until hormone regulatory signals cause them to move to the cell membrane for exocytosis-mediated release into the bloodstream (Fig. 2A). Their common structural and functional features gave rise to the APUD (amine precursor uptake and decarboxylation) theory by Pearse in 1974 [8]. There are two subfamilies of neuroendocrine cells: epithelial neuroendocrine cells, derived mainly from the embryonic endoderm, and non-epithelial neuroendocrine cells (paraganglia, also called “paraneurons”), derived from the embryonic neuroectoderm. Epithelial neuroendocrine cells include pituitary adenohypophysial cells, parathyroid cells, parafollicular C-cells, pancreatic islet cells, and the cells of the so-called “dispersed neuroendocrine system” that populate the mucosa of the respiratory and gastrointestinal tracts from the nose and mouth to the rectum, as well as the genitourinary system [7]; these cells are also present in the skin where they are known as Merkel cells. Paraganglia are present in soft tissue from the base of the skull to the bottom of the vertebral column; the largest form the adrenal medulla bilaterally, and they include the para-aortic paraganglia, the carotid body, the jugulotympanic paraganglion (also called “glomus jugulare”), the organ of Zuckerkandl, and paraganglia of the lungs and porta hepatis [9]. All neuroendocrine cells express a number of proteins that are characteristic of their differentiation, including neural cell adhesion molecule that is recognized by the antibody CD56, neuron-specific enolase, synaptophysin, PGP 9.5, chromogranins, and the transcription factor insulinoma-associated protein-1 (INSM1); several of these are not specific for neuroendocrine cells and only chromogranins and INSM1 represent specific biomarkers of neuroendocrine differentiation (Fig. 2B, C) [10] as they are only also expressed by neural tumors [11]. In addition to these common biomarkers, most epithelial neuroendocrine cells express keratins (that are not present in paraganglia) and specific transcription factors and hormones that allow cell type characterization, whereas paraganglia express the transcription factor GATA3 and tyrosine hydroxylase, a critical enzyme early in the biosynthetic pathway of catecholamines.
Fig. 1: The Families of Neuroendocrine Cells. 
These highly specialized hormone-secreting cells are divided into two families, the epithelial group that usually express keratins and the paraganglia that derive from the sympathetic and parasympathetic autonomic nervous system and usually do not express keratins. Epithelial neuroendocrine cells can comprise a neuroendocrine organ, such as the pituitary or parathyroid, or they can be dispersed throughout non-endocrine or non-neuroendocrine tissues. Each group of epithelial neuroendocrine cells is composed of specific cell types that have distinct structure and function.
Fig. 2: Neuroendocrine features of pituitary neuroendocrine tumors. 
A The ultrastructure of this PitNET from a patient with acromegaly illustrates the neuroendocrine features of adenohypophysial cells. The tumor cell cytoplasm contains rough endoplasmic reticulum (*) for hormone synthesis, a prominent Golgi complex (G) where hormone is packaged into forming secretory granules, and numerous membrane-bound secretory granules that store hormone for release into the extracellular space. B The tumor cells show cytoplasmic chromogranin reactivity. C Nuclear INSM1 positivity is a feature of pituitary neuroendocrine tumors.


Pituitary adenohypophysial cells are a complex family of neuroendocrine cells that, like other related cells, express transcription factors that determine their ability to synthesize specific hormones [12]. Studies of cytodifferentiation have demonstrated three major families of pituitary adenohypophysial cells, with six terminally differentiated cell types (Fig. 3). In the mature gland, there is evidence to suggest that transdifferentiation occurs physiologically between the four cell types of the PIT1 family; it is unclear if transdifferentiation occurs between corticotrophs, gonadotrophs and the PIT1-lineage somatotrophs, lactotrophs, mammosomatotrophs, and thyrotrophs. In general, PitNETs are classified as originating from these various cell types, but in addition, morphologic variants of these mature cells have different patterns of granulation, and some tumors show differentiation along a lineage without the features of terminally differentiated mature cell phenotypes (Fig. 3). Null cell tumors do not show evidence of adenohypophysial lineage differentiation; with improved techniques they represent as low as 0.6% of all adenohypophysial tumors [13], consistent with the initial proposal that with advances in knowledge, they would all but disappear [14] and now their continued existence has been questioned [15]. Mixed tumors also occur; they may be of a single lineage, such as the mixed tumors of somatotrophs and lactotrophs that may represent divergent differentiation of a single cell type, or they may be synchronous distinct tumors of different lineages [16]. There are also unusual plurihormonal tumors that show mixed lineage [1, 17]. Very rare and unusual pituitary tumors of primitive adenohypophysial cells (composed of a mixture of adenohypophysial neuroendocrine cells, primitive Rathke’s cleft cells, and primitive folliculostellate cells) are known as pituitary blastomas [18].

Adenohypophysial hormone-secreting cells differentiate along three lineages to form multiple mature cell types. Each mature cell type is associated with at least one tumor type, but several cell types give rise to multiple tumor variants. In addition, there are tumors composed of cells that lack terminal differentiation; some of these show lineage differentiation but others, most commonly those classified as null cell tumors, lack evidence of any lineage specificity.
Terminologies in classification of pituitary NETs
Tumors composed of adenohypophysial hormone-secreting cells have been classified as “adenomas” since their early descriptions in the late 19th century and early 20th century. This terminology has been applied even to tumors that extend upwards to compress the optic chiasm and brain, and to those that infiltrate dura and invade laterally to involve the cavernous sinuses, as well as to tumors that penetrate the sphenoid bone, clivus, and/or paranasal sinuses. The term “carcinoma” has been restricted to PitNETs that show evidence of distant metastasis to cervical lymph nodes, lung, liver and bone, and/or discontinuous cerebrospinal fluid dissemination within the cranium or spinal canal [1, 2]. This represents a divergence from some other tumor classification schemes in which invasion represents the defining characteristic of a malignancy, for example, epithelial neoplasms including squamous and basal cell carcinomas, breast carcinomas, and intestinal malignancies.
By definition, an “adenoma” is a “benign tumor of glandular epithelial cells.” The definition of “benign” is “a disease that is not harmful in effect.” The Merriam-Webster dictionary defines benign as “of a mild type or character that does not threaten health or life,” as “having no significant effect.”
To address the discrepancy between benign behavior and the morbidity of patients with invasive tumors that cannot be resected surgically, efforts were made to classify some tumors as “atypical adenomas” [19]. However, the criteria for this diagnosis suffered from a lack of reproducibility, and this terminology has since been abandoned [2].
In other sites, tumors of neuroendocrine cells have had varying terminologies applied and they have undergone changes over time. In the pancreas, “islet cell adenoma” was replaced by “islet cell tumor” when it became clear that they can behave aggressively; subsequent studies that showed origin of some of these tumors from neuroendocrine cell precursors in pancreatic ducts rather than in islets resulted in a further change to “pancreatic neuroendocrine tumors” (pNETs or PanNETs) [19, 20]. Oberndorfer initially classified small bowel NETs as “carcinoid” tumors because they looked “carcinoma-like” [21]; once their behavior was clarified as potentially malignant, the term “carcinoid” was mostly restricted to the clinical syndrome of serotonin excess that is a feature of the enterochromaffin (EC) cell neoplasms, and these tumors assumed the terminology of intestinal NETs along with their counterparts, gastric, and rectal NETs. In the lung, the terminology of “typical and atypical carcinoid tumors” remains embedded in the literature [22], but is likely to change over time [23].
Molecular analysis combined with careful clinical studies have shown that well-differentiated NETs have unique genetic and epigenetic alterations that are not infrequently germline, giving rise to familial syndromes such as multiple endocrine neoplasia types 1, 2, 4, and 5, von Hippel Lindau syndrome, tuberous sclerosis, neurofibromatosis syndromes, and the succinate dehydrogenase (SDH)-related [24,25,26,27]. In contrast, poorly differentiated neuroendocrine neoplasms (NENs), including high-grade malignancies in the lung and upper respiratory tract, pancreas and gut, breast, and genitourinary system, are more closely related to non-endocrine carcinomas, both genetically and clinically [28, 29] with some exceptions in the lung [30].
To address the issues of terminology, the World Health Organization (WHO) proposed the implementation of a unified approach to the classification of neuroendocrine neoplasia at all body sites. The proposal includes the use of the term “NEN” to describe all members of this large family of proliferative lesions of neuroendocrine cells [23]. The term “neuroendocrine carcinoma” (NEC) was proposed for the aggressive poorly differentiated high-grade malignancies; in contrast, the well-differentiated and generally low-grade neoplasms were classified as “NETs” in keeping with the terms in place for gastro-entero-pancreatic lesions.
The term “tumor” is indeed problematic. Etymologically derived from the Latin words “tumere,” meaning “to swell” and/or “tumor,” meaning “swelling,” this word has been used since the 16th century to mean a mass that can be inflammatory, neoplastic, or vascular. However, in modern medical vernacular, it has taken on the context of neoplasia, and in the case of “NET,” it has a specific connotation, implying a neoplasm composed of well-differentiated neuroendocrine cells, usually with low proliferative activity [23]. There is no specific indication of benign or malignant classification, as it is recognized that these lesions may show a wide spectrum of behaviors, including a potential for local invasion and metastasis.
In 2016, to address the clinical need for recognition of the morbidity caused by invasive and aggressive pituitary “adenomas” and their metabolic disturbances, and anticipating a convergence of classifications of NETs at various body sites, the International Pituitary Pathology Club proposed a change in nomenclature for tumors of hormone-secreting adenohypophysial cells from “adenoma” to “NET” [31]; in parallel with the pancreatic abbreviation “PanNETs,” the term was abbreviated to “PitNETs.” This terminology is consistent with the WHO proposal. It recognizes these pituitary neoplasms as members of the neuroendocrine family. The term “tumor” replacing adenoma provides for the spectrum of behavior from indolent lesions to those with significant hormonal impact and/or invasive growth, thus recognizing the morbidity of aggressive tumors without implying overt malignancy. It also avoids the contradiction when a pituitary tumor develops a metastasis, which, although rare, usually follows a prior diagnosis of “adenoma.”
Another important aspect related to terminology and tumor classification is the categorization of histological tumor subtypes based on clinical and biochemical features. The term “pituitary NET” is a classification for the family of tumors, and each tumor subtype should be classified based on cell type, e.g., corticotroph, lactotroph, somatotroph, mammosomatotroph, thyrotroph, or gonadotroph tumor (Fig. 3). The clinical presentation is important. Most somatotroph and lactotroph tumors are clinically functioning and the classical presentation of a corticotroph tumor is Cushing disease; however, some of these differentiated hormone-producing tumors are clinically “silent.” In contrast, the vast majority of gonadotroph tumors are clinically silent. Clinicians should be aware that among tumors that are clinically classified as nonfunctioning, the majority are gonadotroph tumors that tend to be indolent, but there are also tumors that are morphologically classified as silent corticotroph, silent somatotroph, and other silent tumors of PIT1 lineage, and very rarely they can be classified as true null cell tumors with no evidence of pituitary lineage differentiation.
Unlike in other sites, detailed structure–function correlations in pituitary tumors have shown that there are structural variants of hormone-producing cell types that are clinically relevant. For instance, the clinical entity known as acromegaly is not a single disease [32]; densely and sparsely granulated variants of somatotroph tumors have distinct clinical and radiologic features and different responses to therapy, and several other tumors of PIT1 lineage can also manifest with acromegaly. Similarly, a TSH-producing PitNET is not always a thyrotroph tumor, since both mature and poorly differentiated plurihormonal PIT1-lineage tumors can also result in central hyperthyroidism [33, 34]. A substantial advance in knowledge has been achieved in PitNETs by clinicopathologic and genomic studies that have applied rigid criteria to distinguish histological tumor subtypes.
Epidemiology
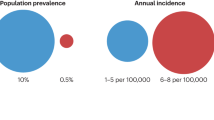
Like other NETs that have been increasing in incidence [35], PitNETs that were once considered to be rare are increasingly diagnosed. They are frequently identified incidentally at autopsy or on routine radiologic examination for other reasons. Autopsy studies have reported a prevalence as high as 22.5 [36] and 27% [37] of cases. Approximately 20% of “normal” pituitary glands harbor an incidental radiologically detectable lesion measuring 3 mm or more in diameter [4, 38]. The majority of these incidental findings are clinically nonfunctioning pituitary tumors, but even these “asymptomatic” tumors can cause hypopituitarism [39] while others are prolactinomas that cause undiagnosed clinical symptomatology [40, 41]; occasionally they are somatotroph or corticotroph tumors causing clinically undiagnosed acromegaly and Cushing disease, respectively.
In several population studies from the last 15 years, the prevalence of clinically diagnosed pituitary tumors ranges from ~78 to 116 cases per 100,000 people [6, 42,43,44]; one study from a Swedish group reported an incidence of 3.9/100, 000 people [45]. Data from the Central Brain Tumor Registry of the United States from 2011 to 2015 show pituitary tumors as 17.5% of reported brain tumors; they estimate an incidence rate of 4.12 sellar tumors/100,000 population per year, but this figure is clearly low, since these statistics are based only on surgically resected tumors [46]. In a recent Korean nationwide population-based study, the annual incidences per 100,000 population were estimated to be 3.5 for nonfunctioning pituitary tumors, 1.6 for lactotroph tumors, 0.5 for somatotroph tumors, and 0.2 for corticotroph or thyrotroph tumors [47]. Independent of country-specific differences, epidemiologic studies considered together show that clinically apparent PitNETs are increasing in both annual incidence (between 3.9 and 7.4 cases per 100,000) and prevalence (76–116 cases per 100,000 population), the latter corresponding roughly to 1 case per 1000 people, with nonfunctioning tumors and prolactinomas as the most frequent subtypes [48].
Prolactinomas are consistently the most common type of PitNET [4, 6, 42,43,44,45, 48]; they are usually treated medically [49,50,51,52] and most are therefore not captured in cancer or surgical statistics, so their exact incidence is unknown. In one prevalence study, surgical resection was performed for 56% of patients with clinically diagnosed PitNETs [6]. The relative frequency of the various tumor types resected surgically shows that more than a third are hormonally inactive mass lesions of SF1 lineage [53, 54]. About 30% are of PIT1 lineage and more than half of those give rise to growth hormone excess, while ~15% are TPIT lineage tumors [6, 42, 49, 52, 54, 55].
Prognostic biomarkers in pituitary NETs
The prognosis of a patient with a PitNET depends on multiple factors, which are collectively described as “aggressiveness.” The relevant parameters that define aggressiveness include hormone secretory activity, tumor size, site, extent of invasiveness, and rate of growth into surrounding structures as well as refractoriness to therapy. Together, these parameters influence the success of surgical resection, metabolic disorders caused by the tumor, along with other intrinsic tumor characteristics that collectively impact disease-specific morbidity.
While the vast majority of prolactinomas are clearly not aggressive, some are resistant to dopamine agonist therapy and the rare very large tumors in men, can be more aggressive, not only because they are larger at presentation but possibly because of altered expression of genes that are mediated by estrogen receptor signaling [56]. In the following paragraphs, we review the evidence that is available to help define aggressiveness based on the extent of disease and tumor type.
Of patients who undergo surgery, the percent that have persistent disease varies depending on the size, invasiveness of the tumor, and extent of resection [57]. Recurrence after surgery has been reported to vary from 0 to 40% in those with 5 or more years of follow-up; the variability is often due to the duration of follow-up. In one series, progression occurred in 27.8% of patients and the mean time to progression was 23.7 months (range, 3.7–52.4 months), both associated with tumor size and extent of resection [58]. Neurosurgeons have defined the extension of tumors into adjacent neuroanatomical regions (Fig. 4) such as the degree of lateral extension into the cavernous sinus (Knosp classification) [59] and also the degree of extra-sellar and vertical extension into suprasellar regions (Hardy classification) [60]. Many patients with Knosp grade 3 or higher cavernous sinus invasion eventually require multimodal therapies including repeat operations and/or radiotherapy [61]. Despite efforts to predict cavernous sinus invasion on magnetic resonance imaging (MRI), the precise confirmation of invasion frequently requires surgical and/or histological examination. Clearly there is a need for better biomarkers to help predict invasiveness [62].

Contrast enhanced T1-weighted MRI of sella of a locally invasive macrotumor that was associated with visual loss and weight gain. On investigation, the ACTH level was 237 pg/mL (normal, <50) and morning cortisol 28 μg/dL (normal, 4–22), providing a biochemical diagnosis of Cushing disease. The tumor surrounds the pituitary gland, which is seen as an ill-defined focus of enhancement in the middle of the sella, and fills both cavernous sinuses (both Knosp grade 4), with suprasellar extension through the oculomotor triangle into the basal cistern just above the right cavernous sinus, involvement of the clivus, and invasion into the sphenoid sinus. A diagnosis of pituitary carcinoma cannot be assigned because of the absence of any noncontiguous metastatic tumor. This highly invasive tumor shows the incoherence of diagnosis as a grade I “adenoma” in the face of such aggressive clinical behavior.
Surgical remission varies by tumor type [1, 63]. In a surgical series that included multiple hormonally active tumor types, at a median follow-up of 56 months, tumor recurrence was 0, 11, and 14% for GH-, ACTH-, and PRL-secreting tumors [64]. In another study, the recurrence-free survival at 10 years was 78.2% in acromegalic patients, 68.1% in prolactinomas, 74.3% in Cushing disease, 70.3% in TSH-secreting tumors and 75.3% in clinically hormonally inactive tumors [65]. In yet another series, corticotroph tumors had a higher risk of recurrence than other tumor types [66].
The Knosp classification has been reported to correlate with surgical outcomes in GH tumors [67]. When surgery is performed by an experienced surgeon, remission is >85% for microtumors <1 cm [68,69,70] but only 40–66% for macrotumors [69,70,71,72]. Because GH-secreting tumors can invade into sellar dura and/or the cavernous sinus at the time of diagnosis, the overall control rate with strict hormonal criteria is 50–80% [67, 73,74,75]. Occult dural foci of tumor may provide the basis for persistent acromegaly after gross total resection and a clear postoperative MRI. Many studies report the outcome of transsphenoidal surgery as the first-line treatment; the impact of preoperative medical therapy for GH-secreting tumors to address comorbidities and reduce soft tissue swelling to facilitate the operative approach remains the subject of debate.
For ACTH-secreting tumors, reported remission rates vary from 60 to 90%, but up to 15–20% eventually recur [76,77,78,79]. Pivonello et al. [80] reviewed 74 studies published between 1976 and 2014 involving 6134 patients with Cushing disease and a mean follow-up duration of 64.3 months. In their review, they found that the overall initial remission rate ranged from 25 to 100%, with a mean remission rate of 77.8% (median, 78.7%); the recurrence rate ranged from 0 to 65.6%, with a mean recurrence rate of 13.2% (median, 10.6%). The remission rates in microtumors ranged from 48.7 to 100% (mean, 82.1%; median, 85.7%), whereas in macrotumors, they ranged from 30.8 to 100% (mean, 62.3%; median, 64.1%). The recurrence rates in patients with microtumors and macrotumors were 0–36.4% (mean, 11.7%; median, 10.9%) and 0–59% (mean, 18.8%; median, 13.9%), respectively. Although en bloc resection achieves the best rates of remission (98.5%), it applies only to smaller tumors not invading dura or other parasellar structures [81]. Dickerman and Oldfield noted the clinical importance of occult dural invasion, i.e., invasion of the dura or cavernous sinus that is not evident on imaging studies and is not obvious to the surgeon, as the basis of recurrence or persistent tumor and endocrinopathy in their series of Cushing disease [82].
After surgery, the probability of recurrence of apparently completely resected tumors and the time to clinically relevant regrowth of known residual tumor correlate with radiologic tumor doubling-time [83]. However, there is controversy about the role of proliferation markers including mitoses (conventional or phosphohistone-H3-assisted count), Ki-67 labeling index, and p53 staining [84, 85].
While transsphenoidal surgery is the initial treatment of choice for the great majority of large pituitary tumors, many cases of functional tumors cannot be hormonally controlled, and the recurrence rate of nonfunctioning tumors is high. In tumors that cannot be fully resected, the prognosis depends on multiple factors that mediate tumor responsiveness to other therapeutic modalities. These include therapies used to control hormone hypersecretion as well as those intended to target structural tumor growth. Patients with acromegaly and residual tumor postoperatively are more likely to respond to first-generation somatostatin analogues if their disease is due to a densely granulated somatotroph or mammosomatotroph tumor than if they have a sparsely granulated somatotroph tumor [86,87,88,89,90]. It has been reported that patients with persistent or recurrent Cushing disease are more likely to respond to pasireotide if they have a tumor with a USP8 mutation [91], a molecular alteration that is more frequent in densely granulated microtumors [91, 92], again emphasizing the importance of tumor histotype in determining therapeutic responsiveness.
Among patients with clinically nonfunctioning tumors, classification by cell type is equally important [13]. Among clinically nonfunctioning tumors, gonadotroph tumors are more indolent whereas true null cell tumors and silent corticotroph tumors are more likely to behave aggressively [13, 93,94,95]. For example, in one report of silent corticotroph tumors with a follow-up longer than 5 years, 31% recur (CI 13–38%) [96]. Approximately 50% of patients with detectable postoperative residual tumor develop progression within a 5-year period [97,98,99]; however, the most common gonadotroph tumors tend to grow slowly, whereas silent corticotroph tumors, while not always reported to have a higher recurrence rate [96], have been shown in most studies to be more proliferative and invasive, resulting in relatively more frequent compressive features with visual field loss, hypopituitarism, and other symptoms and signs of tumor progression [100,101,102,103,104].
Certain tumor types are considered to be inherently aggressive. The so-called “poorly differentiated” PIT1-lineage tumors (formerly known as “silent subtype 3 pituitary adenomas”), which are thought to lack terminal PIT1-directed cell maturation, have been associated with frequent disease progression in three clinical series [33, 105, 106]. Crooke cell tumors have been well-recognized for their adverse biology [107,108,109]. In the group of somatotroph tumors, sparsely granulated tumors are more aggressive than densely granulated tumors; they present with larger and more invasive lesions [87, 89, 110,111,112]. Among prolactin-producing tumors, acidophil stem cell tumors have a higher risk of aggressive disease [113, 114] and are often resistant to medical therapy [115]. TSH-secreting tumors historically showed more invasive behavior and were more difficult to fully resect [116]; however, this may relate to delays in diagnosis, and inappropriate treatment with thyroid ablation that resulted from misdiagnosis as primary hyperthyroidism.
It has been estimated that of pituitary tumors requiring surgery, some 15% can be considered as highly aggressive based on their surgical, radiological, and pathological characteristics [117, 118]. Such patients may be resistant to both conventional radiotherapy and radiosurgery. Newer medical agents adopted in the broader field of NETs are beginning to make their way into the therapeutic armamentarium for a subset of unresectable or especially aggressive PitNETs. These include the mTOR inhibitor everolimus [119, 120] as well as the DNA methylation inhibitor temozolomide [121,122,123,124]. In other tissues, responsiveness to temozolomide has been shown to be predicted by the level of expression of O6-methylguanine-DNA methyltransferase but it is unclear if this is useful in the case of PitNETs [122, 123, 125, 126]. Equally as important, the optimization of therapeutic responsiveness of temozolomide-based therapy through the combined use with capecitabine has resulted in the now widely adopted CAP/TEM regimen in NET management [127, 128]; it is expected that CAP/TEM will similarly be used for temozolomide monotherapy escape or resistant PitNETs [128,129,130]. Peptide radio-receptor therapy with 177lutetium (177Lu)-based protocols represents another major advance in NET management that will likely extend to use in PitNETs [129, 131]. Such NET management strategies, alone or in sequence, will prove to be critically important in directing their extended use for aggressive PitNETs.
These data show the importance of determining a dynamic multifactorial clinicopathologic risk stratification. This has been attempted [57]; however, it is important to distinguish tumor grade from risk score and tumor stage. It is important to apply grading based not only on proliferation but also on differentiation, and there is clearly a need for appropriate staging of PitNETs to allow the determination of prognosis based on surgical resectability. The prognosis for patients with inoperable disease is dependent on response to medical therapy that is mainly a reflection of cell type and subtype.
Genetic predisposition and pathogenesis-related biomarkers
Like other NETs, PitNETs are members of several families of tumors associated with a genetic predisposition. They form one of the three “P”s of multiple endocrine neoplasia type 1 (MEN1), originally described by Wermer [132]. This autosomal dominant disorder with incomplete penetrance is due to inheritance of a mutant copy of the MEN1 gene that encodes menin. Approximately half of the affected patients develop a PitNET; the most common types produce prolactin and/or GH [4, 133,134,135] and poorly differentiated tumors of PIT1 lineage have been reported in patients with MEN1 [33]. The tumors may be multiple and multicentric [134, 136], but this can be hard to prove. Patients with MEN1 generally present at a younger age than those with sporadic tumors [135]. Other manifestations of MEN1 include parathyroid and pancreatic neoplasms as well as frequent adrenal cortical tumors [137] and, more recently, breast cancer has been shown to be more frequent in patients with MEN1 [138].
Defects in CDKI genes are responsible for an MEN1-like syndrome known as MEN4. Patients have mutations in the CDKN1B/p27Kip1 gene [139, 140] and the CDKN2C/p18INK4c gene [141, 142]. MEN4 is characterized by parathyroid proliferations, PitNETs, and pNETs as well as small intestinal NETs, lymphoma, and breast cancer [143].
Cases of MEN1-like acromegaly associated with hyperparathyroidism and pNETs have been described in patients with germline mutation of CDC73 [144, 145]. Germline pathogenic MAX variants have been associated with PIT1-lineage tumors along with other endocrine and non-endocrine tumors in a syndrome proposed as MEN5 [27].
A small number of patients with pituitary GH-secreting tumors, most often those presenting at a young age and associated with gigantism, have Carney’s complex [146], an autosomal dominant disorder associated in around 50% of cases with germline mutations in the PRKAR1Aα gene that encodes the PKA regulatory subunit 1Aα, causing unrestrained cAMP signaling [147, 148]. The syndrome is characterized by spotty pigmentation due to nevi affecting mucosal surfaces and the lips, malignant melanotic nerve sheath tumors, cardiac and other myxomas, and endocrine tumors including not only pituitary tumors but also bilateral pigmented nodular adrenocortical disease and testicular tumors [146].
Mutations in genes encoding the various components of the SDH complex are implicated in several types of familial neoplasia [149] including pheochromocytomas and paragangliomas, gastrointestinal stromal tumors, and renal carcinomas as well as rare pituitary tumors [150].
Lynch syndrome is a well-recognized autosomal dominant familial cancer predisposition syndrome that increases the risk of many types of cancer, mainly colorectal carcinomas but also tumors of the stomach, small intestine, liver, gallbladder, upper urinary tract, brain, skin, and adrenal cortex; women with this disorder have a high risk of ovarian and endometrial carcinomas. It is due to mutation in one of the genes encoding mismatch repair proteins (MLH1, MSH2, MSH6, or PMS2). However, recently NETs, including pancreatic and pituitary tumors, have also been reported in patients with this disorder [151, 152].
In some cases, familial transmission is unique to pituitary tumors without other associated NETs. Predisposition to develop pituitary GH-producing tumors without other endocrine tumors was originally described as the isolated familial somatotropinoma syndrome [153]. Subsequent studies identified predisposition for other types of pituitary tumors as well, and the nomenclature was altered to the familial isolated pituitary adenoma syndrome [154]. The pituitary tumors in these families arise at a younger age and are larger than tumors in matched sporadic pituitary tumor cohorts. This syndrome is associated in around 20% of cases with germline mutations in the AIP gene that encodes aryl hydrocarbon-interacting protein (AIP), which functions as a tumor suppressor [154,155,156]. AIP is a chaperone protein that modulates transcription of the aryl hydrocarbon receptor, which was originally described to mediate toxicological and carcinogenic dioxin effects [157, 158].
A rare form of early childhood X-linked acrogigantism attributed to Xq26 microduplications and GPR101 mutation [159] may primarily predispose to hyperplasia but somatotroph and/or mammosomatotroph PitNETs have also been described [160].
Sporadic tumors rarely have mutations in the genes that are implicated in these familial syndromes [1, 161]. Instead, mutations have only been identified only in small subsets of tumors. For example, a subset of somatotroph tumors have activating mutations of GNAS resulting in tumors that have high cyclic AMP levels and responsiveness to somatostatin analogues; several studies have shown that this mutation is most often seen in tumors with a densely granulated somatotroph phenotype [32, 162] but not all densely granulated tumors harbor GNAS mutations and the correlation is not perfect [110, 111]; histopathology remains the best predictor of therapeutic responsiveness [90]. Some corticotroph tumors have mutations of USP8, a gene encoding a member of the ubiquitin protease family and is thought to alter EGFR signaling; this feature is predictive of pasireotide response [91] and tends to be identified in small densely granulated corticotroph tumors [91, 92, 163, 164]. Interestingly, a recent study has identified ATRX mutations in aggressive pituitary tumors, mostly of corticotroph type, including those with metastases [165], similar to pNETs [166] and paragangliomas [167]. Nevertheless, the majority of sporadic PitNETs lack recurrent mutations, showing only variable genomic instability with copy-number variations [168,169,170,171,172], and like small bowel NETs [173], epigenetic dysregulation has been implicated in their pathogenesis [174].
Clinical management: the need for centers with sustained experience
Endocrine tumors are complex and require broad multidisciplinary expertise. Unlike other solid tumors, they produce a number of hormonal disturbances that have significant importance to the patient and that often are as important or perhaps more so than tumor growth. In the case of PitNETs, the clinical team includes endocrinologists, neuroradiologists, neurosurgeons, pathologists, medical and radiation oncologists, specialized nurses, and psychosocial experts who are knowledgeable about the emotional disorders that are common in patients with these tumors [175,176,177]. The need for psychosocial care in oncology is now becoming widely recognised, but the recognition of the hormonal aspects of endocrine neoplasia adds another dimension to this problem. A strong and integrated multidisciplinary team is required to allow for personalized approaches to care and ongoing support.
Throughout the various areas of medicine, subspecialisation has been proven to result in significant quality improvement, error reduction, and cost avoidance [178,179,180,181,182,183,184,185,186,187,188,189,190]. Data show consistently that all members of the care team have better outcomes with increasing numbers of patients. Experts are able to create standards based on evidence from their practices. For example, pathologists who see many cases of pituitary tumors each year have formulated a synoptic report that provides a standard approach to morphologic diagnosis and structure–function correlations [191]. Although the designation of high-volume centers as “Centers of Excellence” has been proposed [192], as yet no criteria have been created for such a designation, nor any external assessment protocols that provide objective measures of “excellence.”
PitNETs as a model for other NETs
As we have shown in the previous paragraphs, the careful categorization of PitNETs into different morpho-functional subtypes according to cell lineage and specific structural properties of neoplastic cells leads to a classification with important predictive and prognostic implications. In fact, such a degree of insight into structure–function–behavior relationships has not been reached for NETs arising in other organs. Nevertheless, it should be recognized that, in almost every anatomical site in which neuroendocrine cells occur, except for thyroid and parathyroid where only a single neuroendocrine cell type is recognized, NENs exhibit a wide morpho-functional spectrum, reflecting and sometimes amplifying the diversity of normal neuroendocrine cells to which they are related.
In particular, it is known that gastrointestinal and pNETs include many different subtypes, defined according to cell morphology and hormone production [193]. However, in the current WHO classifications, these features are only taken into account for functioning PanNETs [2, 194], mainly in relation to the clinical syndromes associated with these neoplasms, whereas the core of the classification is based on the degree of histological differentiation and the proliferative grade. In the small bowel, the pioneering work of Swedish investigators identified more than 20 different cell types with distinct hormone products and ultrastructure in the late 1970s and early 1980s [195]; this work has been largely ignored, likely because the vast majority of tumors are EC cell serotonin-producing lesions that can potentially cause the carcinoid syndrome and its sequelae. It is unclear why EC cells alone, from among the many others scattered throughout the ileum, give rise to tumors. The lungs have at least three neuroendocrine cell types known to produce several peptides, including bombesin, calcitonin, and calcitonin-gene-related peptide, as well as serotonin, and although the hormonal profiles of pulmonary NETs have been investigated [196], this subject has been disregarded, possibly because of a suggested lack of clinical impact. The same is true also for other rare anatomical sites including the upper respiratory tract, genitourinary tract, and others. In general, the field of NETs seems to suffer from an under-appreciation of the clinical significance of the cells of origin and their specific alterations in tumors. The very few exceptions are represented by the work identifying the four cell types in duodenal NETs [197], publications showing that L cell rectal NETs have a distinct morphology and more indolent behavior than non-L cell rectal NETs [198,199,200], and data reporting ectopic hormone secretion in lung [201, 202] and pNETs [201, 203,204,205]. Lessons learned from PitNETs should prompt a re-evaluation of the significance of hormone profiles and careful morphological examination of specific cell types in NETs of other body sites. Documentation of tissue-specific transcription factors and hormone profiles can advance the diagnostic management of NETs, improve the diagnosis of metastatic disease from an unknown primary site [206] and provide biomarkers for clinical surveillance. Moreover, this information can also provide useful tools for the therapeutic choices and prognostic evaluation of patients with these tumors [207, 208].
If the pituitary is to be used as the paradigm of neuroendocrine neoplasia, the field must also align with other NETs in more than just nomenclature. Indeed, the concept of pituitary carcinoma, traditionally applied to metastatic lesions, should be relinquished in light of two important considerations. First, the term NEC is a highly specific term, which is applied only to high-grade neoplasms with distinct genetic alterations more akin to those of other epithelial malignancies [23]. Curiously, there are almost no examples of such lesions in the pituitary with the possible exception of rare TP53-mutant carcinomas. Even the so-called “poorly differentiated” tumors are only considered to be poorly differentiated in the context of the complex and elegant system of cytodifferentiation of adenohypophysial cells, but not in the context of high-grade histopathology; this terminology should be reconsidered in light of the WHO classification and perhaps warrants alternate terminology, such as “immature” rather than “poorly differentiated.” The second consideration is that the term PitNET, in analogy with NETs of other anatomical sites, implies a spectrum of behaviors including potential metastatic spread and, as such, does not require a specific term to distinguish locally aggressive and/or metastatic lesions.
Building on the WHO proposal for a common classification for NENs, we propose a common approach to the investigation of structure–function correlations in the endocrinology and pathology of NETs at all body sites as has been done in the pituitary gland.
Conclusions
-
Pituitary adenohypophysial neoplasms are epithelial and neuroendocrine in nature, thus are fully embedded in the concept of neuroendocrine neoplasm.
-
The vast majority are well-differentiated and fall into the category of NETs, which show a spectrum of behavior from indolent to locally and hormonally aggressive, and occasionally metastatic.
-
Recognition of adenohypophysial cell differentiation provides meaningful biological, clinical, prognostic, and predictive information that informs appropriate management.
-
Dynamic risk stratification for patients with PitNETs currently relies on the tumor cell type and subtype, proliferative activity and invasiveness; there is a need to implement grading and staging for these neoplasms to provide a predictive model of tumor behavior.
-
Standardized data collection and synoptic reporting are strongly recommended.
-
A multidisciplinary approach is essential for optimal outcomes.
-
PitNETs represent an advanced model of structure–function correlations that can be used for research in other NETs.
References
Asa SL, Perry A. Tumors of the pituitary gland. AFIP Atlas of tumor and nontumor pathology, Series 5. Fascicle 1. Arlington VA: ARP Press; 2020.
Lloyd RV, Osamura RY, Kloppel G, Rosai J. WHO classification of tumours of endocrine organs, 4th ed. Lyon: IARC; 2017.
Manojlovic-Gacic E, Rostami E, Karavitaki N, Casar-Borota O. Histopathology of parasellar neoplasms. Neuroendocrinology. 2020;110:740–52.
Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, et al. The prevalence of pituitary adenomas: a systematic review. Cancer. 2004;101:613–9.
Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). Bull Johns Hopkins Hosp. 1932;50:137–95.
Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006;91:4769–75.
Asa SL, Mete O. Endocrine pathology: past, present and future. Pathology. 2018;50:111–8.
Pearse AGE. The APUD cell concept and its implications in pathology. Pathol Annu. 1974;9:27–41.
Asa SL, Ezzat S, Mete O. The diagnosis and clinical significance of paragangliomas in unusual locations. J Clin Med. 2018;7:280.
Rosenbaum JN, Guo Z, Baus RM, Werner H, Rehrauer WM, Lloyd RV. INSM1: a novel immunohistochemical and molecular marker for neuroendocrine and neuroepithelial neoplasms. Am J Clin Pathol. 2015;144:579–91.
Ames HM, Rooper LM, Laterra JJ, Eberhart CG, Rodriguez FJ. INSM1 expression is frequent in primary central nervous system neoplasms but not in the adult brain parenchyma. J Neuropathol Exp Neurol. 2018;77:374–82.
Scully KM, Rosenfeld MG. Pituitary development: regulatory codes in mammalian organogenesis. Science. 2002;295:2231–5.
Nishioka H, Inoshita N, Mete O, Asa SL, Hayashi K, Takeshita A, et al. The complementary role of transcription factors in the accurate diagnosis of clinically nonfunctioning pituitary adenomas. Endocr Pathol. 2015;26:349–55.
Kovacs K, Horvath E, Ryan N, Ezrin C. Null cell adenoma of the human pituitary. Virchows Arch [Pathol Anat]. 1980;387:165–74.
Manojlovic-Gacic E, Bollerslev J, Casar-Borota O. Invited review: pathology of pituitary neuroendocrine tumours: present status, modern diagnostic approach, controversies and future perspectives from a neuropathological and clinical standpoint. Neuropathol Appl Neurobiol. 2020;46:89–110.
Mete O, Alshaikh OM, Cintosun A, Ezzat S, Asa SL. Synchronous multiple pituitary neuroendocrine tumors of different cell lineages. Endocr Pathol. 2018;29:332–8.
Tordjman KM, Greenman Y, Ram Z, Hershkovitz D, Aizenstein O, Ariel O, et al. Plurihormonal pituitary tumor of Pit-1 and SF-1 lineages, with synchronous collision corticotroph tumor: a possible stem cell phenomenon. Endocr Pathol. 2019;30:74–80.
de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014;128:111–22.
DeLellis RA, Lloyd RV, Heitz PU, Eng C. Pathology and genetics of tumours of endocrine organs. 3rd ed. Lyon: IARC; 2004.
Klöppel G, In’t Veld PA, Komminoth P, Heitz PhU. The endocrine pancreas. In: Kovacs K, Asa SL, editors. Functional endocrine pathology. Boston: Blackwell Science; 1998. p. 415–87.
Oberndorfer S. Karzinoide tumoren des Dünndarms. Frankf Z für Pathologie. 1907;1:425–32.
Travis WD, Burke AP, Marx A, Nicholson AG. WHO classification of tumours of the lung, pleura, thymus and heart. 4th ed. Lyon: IARC; 2015.
Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol. 2018;31:1770–86.
Lyssikatos C, Fauez FR, Stratakis CA. Familial endocrine tumor syndromes. In: Mete O, Asa SL, editors. Endocrine pathology. Cambridge: Cambridge University Press; 2016. p. 56–70.
Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71.
Turchini J, Cheung VKY, Tischler AS, de Krijger RR, Gill AJ. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology. 2018;72:97–105.
Seabrook AJ, Harris JE, Velosa SB, Kim E, McInerney-Leo AM, Dwight T, et al. Multiple endocrine tumors associated with germline MAX mutations: multiple endocrine neoplasia type 5? J Clin Endocrinol Metab. 2020;106:1163–82.
Konukiewitz B, Jesinghaus M, Steiger K, Schlitter AM, Kasajima A, Sipos B, et al. Pancreatic neuroendocrine carcinomas reveal a closer relationship to ductal adenocarcinomas than to neuroendocrine tumors G3. Hum Pathol. 2018;77:70–9.
Jesinghaus M, Konukiewitz B, Keller G, Kloor M, Steiger K, Reiche M, et al. Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod Pathol. 2017;30:610–9.
Alcala N, Leblay N, Gabriel AAG, Mangiante L, Hervas D, Giffon T, et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat Commun. 2019;10:3407.
Asa SL, Casar-Borota O, Chanson P, Delgrange E, Earls P, Ezzat S, et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer. 2017;24:C5–8.
Asa SL, Kucharczyk W, Ezzat S. Pituitary acromegaly: not one disease. Endocr Relat Cancer. 2017;24:C1–4.
Mete O, Gomez-Hernandez K, Kucharczyk W, Ridout R, Zadeh G, Gentili F, et al. Silent subtype 3 pituitary adenomas are not always silent and represent poorly differentiated monomorphous plurihormonal Pit-1 lineage adenomas. Mod Pathol. 2016;29:131–42.
Pereira BD, Raimundo L, Mete O, Oliveira A, Portugal J, Asa SL. Monomorphous plurihormonal pituitary adenoma of Pit-1 lineage in a giant adolescent with central hyperthyroidism. Endocr Pathol. 2016;27:25–33.
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–72.
Costello RT. Subclinical adenoma of the pituitary gland. Am J Pathol. 1936;12:205–15.
Burrow GN, Wortzman G, Rewcastle NB, Holgate RC, Kovacs K. Microadenomas of the pituitary and abnormal sellar tomograms in an unselected autopsy series. N Engl J Med. 1981;304:156–8.
Elster AD. Modern imaging of the pituitary. Radiology. 1993;187:1–14.
Freda PU, Bruce JN, Khandji AG, Jin Z, Hickman RA, Frey E, et al. Presenting features in 269 patients with clinically nonfunctioning pituitary adenomas enrolled in a prospective study. J Endocr Soc. 2020;4:bvaa021.
Kovacs K, Ryan N, Horvath E, Singer W, Ezrin C. Pituitary adenomas in old age. J Gerontol. 1980;35:16–22.
McComb DJ, Ryan N, Horvath E, Kovacs K. Subclinical adenomas of the human pituitary. New light on old problems. Arch Pathol Lab Med. 1983;107:488–91.
Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol. 2010;72:377–82.
Agustsson TT, Baldvinsdottir T, Jonasson JG, Olafsdottir E, Steinthorsdottir V, Sigurdsson G, et al. The epidemiology of pituitary adenomas in Iceland, 1955-2012: a nationwide population-based study. Eur J Endocrinol. 2015;173:655–64.
Fontana E, Gaillard R. Epidemiology of pituitary adenoma: results of the first Swiss study. Rev Med Suisse. 2009;5:2172–4.
Tjornstrand A, Gunnarsson K, Evert M, Holmberg E, Ragnarsson O, Rosen T, et al. The incidence rate of pituitary adenomas in western Sweden for the period 2001-2011. Eur J Endocrinol. 2014;171:519–26.
Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018;20:iv1–86.
Oh JS, Kim HJ, Hann HJ, Kang TU, Kim DS, Kang MJ, et al. Incidence, mortality, and cardiovascular diseases in pituitary adenoma in Korea: a nationwide population-based study. Pituitary. 2021;24:38–47.
Daly AF, Beckers A. The epidemiology of pituitary adenomas. Endocrinol Metab Clin North Am. 2020;49:347–55.
Wilson CB, Dempsey LC. Transsphenoidal microsurgical removal of 250 pituitary adenomas. J Neurosurg. 1978;48:13–22.
Terada T, Kovacs K, Stefaneanu L, Horvath E. Incidence, pathology, and recurrence of pituitary adenomas: Study of 647 unselected surgical cases. Endocr Pathol. 1995;6:301–10.
Klibanski A, Zervas NT. Diagnosis and management of hormone-secreting pituitary adenomas. N Engl J Med. 1991;324:822–31.
Kovacs K, Horvath E. Tumors of the pituitary gland. Atlas of tumor pathology, second series, Fascicle 21. Washington, D.C.: Armed Forces Institute of Pathology; 1986.
Feldkamp J, Santen R, Harms E, Aulich A, Modder U, Scherbaum WA. Incidentally discovered pituitary lesions: high frequency of macroadenomas and hormone-secreting adenomas—results of a prospective study. Clin Endocrinol. 1999;51:109–13.
Mete O, Cintosun A, Pressman I, Asa SL. Epidemiology and biomarker profile of pituitary adenohypophysial tumors. Mod Pathol. 2018;31:900–9.
Mindermann T, Wilson CB. Age-related and gender-related occurrence of pituitary adenomas. Clin Endocrinol. 1994;41:359–64.
Wierinckx A, Delgrange E, Bertolino P, Francois P, Chanson P, Jouanneau E, et al. Sex-related differences in lactotroph tumor aggressiveness are associated with a specific gene-expression signature and genome instability. Front Endocrinol. 2018;9:706.
Raverot G, Vasiljevic A, Jouanneau E, Trouillas J. A prognostic clinicopathologic classification of pituitary endocrine tumors. Endocrinol Metab Clin North Am. 2015;44:11–8.
Jang JH, Kim KH, Lee YM, Kim JS, Kim YZ. Surgical results of pure endoscopic endonasal transsphenoidal surgery for 331 pituitary adenomas: a 15-year experience from a single institution. World Neurosurg. 2016;96:545–55.
Knosp E, Steiner E, Kitz K, Matula C. Pituitary adenomas with invasion of the cavernous sinus space: a magnetic resonance imaging classification compared with surgical findings. Neurosurgery. 1993;33:610–7.
Hardy J. Transsphenoidal surgery of hypersecreting pituitary tumors. In: Kohler PO, Ross GT, editors. Diagnosis and treatment of pituitary tumors. International Congress Series No. 303. Amsterdam: Exerpta Medica; 1973. p. 179–98.
Tampourlou M, Ntali G, Ahmed S, Arlt W, Ayuk J, Byrne JV, et al. Outcome of nonfunctioning pituitary adenomas that regrow after primary treatment: a study from two large UK centers. J Clin Endocrinol Metab. 2017;102:1889–97.
Mete O, Ezzat S, Asa SL. Biomarkers of aggressive pituitary adenomas. J Mol Endocrinol. 2012;49:R69–78.
Asa SL, Mete O. Immunohistochemical biomarkers in pituitary pathology. Endocr Pathol. 2018;29:130–6.
Santoro A, Minniti G, Ruggeri A, Esposito V, Jaffrain-Rea ML, Delfini R. Biochemical remission and recurrence rate of secreting pituitary adenomas after transsphenoidal adenomectomy: long-term endocrinologic follow-up results. Surg Neurol. 2007;68:513–8.
Mortini P, Barzaghi LR, Albano L, Panni P, Losa M. Microsurgical therapy of pituitary adenomas. Endocrine. 2018;59:72–81.
Asioli S, Righi A, Iommi M, Baldovini C, Ambrosi F, Guaraldi F, et al. Validation of a clinicopathological score for the prediction of post-surgical evolution of pituitary adenoma: retrospective analysis on 566 patients from a tertiary care centre. Eur J Endocrinol. 2019;180:127–34.
Nishioka H, Fukuhara N, Horiguchi K, Yamada S. Aggressive transsphenoidal resection of tumors invading the cavernous sinus in patients with acromegaly: predictive factors, strategies, and outcomes. J Neurosurg. 2014;121:505–10.
Jane JA Jr., Starke RM, Elzoghby MA, Reames DL, Payne SC, Thorner MO, et al. Endoscopic transsphenoidal surgery for acromegaly: remission using modern criteria, complications, and predictors of outcome. J Clin Endocrinol Metab. 2011;96:2732–40.
Starke RM, Raper DM, Payne SC, Vance ML, Oldfield EH, Jane JA Jr. Endoscopic vs microsurgical transsphenoidal surgery for acromegaly: outcomes in a concurrent series of patients using modern criteria for remission. J Clin Endocrinol Metab. 2013;98:3190–8.
Freda PU, Wardlaw SL, Post KD. Long-term endocrinological follow-up evaluation in 115 patients who underwent transsphenoidal surgery for acromegaly. J Neurosurg. 1998;89:353–8.
Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur J Endocrinol. 2005;152:379–87.
Swearingen B, Barker FG, Katznelson L, Biller BM, Grinspoon S, Klibanski A, et al. Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998;83:3419–26.
Antunes X, Ventura N, Camilo GB, Wildemberg LE, Guasti A, Pereira PJM, et al. Predictors of surgical outcome and early criteria of remission in acromegaly. Endocrine. 2018;60:415–22.
Fathalla H, Cusimano MD, Di Ieva A, Lee J, Alsharif O, Goguen J, et al. Endoscopic versus microscopic approach for surgical treatment of acromegaly. Neurosurg Rev. 2015;38:541–8.
Chen CJ, Ironside N, Pomeraniec IJ, Chivukula S, Buell TJ, Ding D, et al. Microsurgical versus endoscopic transsphenoidal resection for acromegaly: a systematic review of outcomes and complications. Acta Neurochir. 2017;159:2193–207.
Alexandraki KI, Kaltsas GA, Isidori AM, Storr HL, Afshar F, Sabin I, et al. Long-term remission and recurrence rates in Cushing’s disease: predictive factors in a single-centre study. Eur J Endocrinol. 2013;168:639–48.
Alahmadi H, Cusimano MD, Woo K, Mohammed AA, Goguen J, Smyth HS, et al. Impact of technique on cushing disease outcome using strict remission criteria. Can J Neurol Sci. 2013;40:334–41.
Yamada S, Inoshita N, Fukuhara N, Yamaguchi-Okada M, Nishioka H, Takeshita A, et al. Therapeutic outcomes in patients undergoing surgery after diagnosis of Cushing’s disease: a single-center study. Endocr J. 2015;62:1115–25.
Brichard C, Costa E, Fomekong E, Maiter D, Raftopoulos C. Outcome of transsphenoidal surgery for Cushing disease: a single-center experience over 20 years. World Neurosurg. 2018;119:e106–17.
Pivonello R, De LM, Cozzolino A, Colao A. The treatment of Cushing’s disease. Endocr Rev. 2015;36:385–486.
Oldfield EH. Cushing’s disease: lessons learned from 1500 cases. Neurosurgery. 2017;64:27–36.
Dickerman RD, Oldfield EH. Basis of persistent and recurrent Cushing disease: an analysis of findings at repeated pituitary surgery. J Neurosurg. 2002;97:1343–9.
Monsalves E, Larjani S, Loyola GB, Juraschka K, Carvalho F, Kucharczyk W, et al. Growth patterns of pituitary adenomas and histopathological correlates. J Clin Endocrinol Metab. 2014;99:1330–8.
Grimm F, Maurus R, Beschorner R, Naros G, Stanojevic M, Gugel I, et al. Ki-67 labeling index and expression of p53 are non-predictive for invasiveness and tumor size in functional and nonfunctional pituitary adenomas. Acta Neurochir. 2019;161:1149–56.
Salehi F, Agur A, Scheithauer BW, Kovacs K, Lloyd RV, Cusimano M. Ki-67 in pituitary neoplasms: a review-part I. Neurosurgery. 2009;65:429–37.
Ezzat S, Kontogeorgos G, Redelmeier DA, Horvath E, Harris AG, Kovacs K. In vivo responsiveness of morphological variants of growth hormone-producing pituitary adenomas to octreotide. Eur J Endocrinol. 1995;133:686–90.
Bhayana S, Booth GL, Asa SL, Kovacs K, Ezzat S. The implication of somatotroph adenoma phenotype to somatostatin analog responsiveness in acromegaly. J Clin Endocrinol Metab. 2005;90:6290–5.
Heck A, Ringstad G, Fougner SL, Casar-Borota O, Nome T, Ramm-Pettersen J, et al. Intensity of pituitary adenoma on T2-weighted magnetic resonance imaging predicts the response to octreotide treatment in newly diagnosed acromegaly. Clin Endocrinol. 2012;77:72–8.
Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol. 2012;76:96–102.
Ezzat S, Caspar-Bell GM, Chik CL, Denis MC, Domingue ME, Imran SA, et al. Predictive markers fo postsurgical medical management of acromegaly: a systematic review and consensus treatment guideline. Endocr Pr. 2019;25:379–93.
Hayashi K, Inoshita N, Kawaguchi K, Ardisasmita AI, Suzuki H, Fukuhara N, et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur J Endocrinol. 2016;174:213–26.
Ma ZY, Song ZJ, Chen JH, Wang YF, Li SQ, Zhou LF, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015;25:306–17.
Balogun JA, Monsalves E, Juraschka K, Parvez K, Kucharczyk W, Mete O, et al. Null cell adenomas of the pituitary gland: an institutional review of their clinical imaging and behavioral characteristics. Endocr Pathol. 2015;26:63–70.
Almeida JP, Stephens CC, Eschbacher JM, Felicella MM, Yuen KCJ, White WL, et al. Clinical, pathologic, and imaging characteristics of pituitary null cell adenomas as defined according to the 2017 World Health Organization criteria: a case series from two pituitary centers. Pituitary. 2019;22:514–9.
Haddad AF, Young JS, Oh T, Pereira MP, Joshi RS, Pereira KM, et al. Clinical characteristics and outcomes of null-cell versus silent gonadotroph adenomas in a series of 1166 pituitary adenomas from a single institution. Neurosurg Focus. 2020;48:E13.
Fountas A, Lavrentaki A, Subramanian A, Toulis KA, Nirantharakumar K, Karavitaki N. Recurrence in silent corticotroph adenomas after primary treatment: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2018;10:1210.
Batista RL, Trarbach EB, Marques MD, Cescato VA, da Silva GO, Herkenhoff CGB, et al. Nonfunctioning pituitary adenoma recurrence and its relationship with sex, size, and hormonal immunohistochemical profile. World Neurosurg. 2018;120:e241–46.
Dekkers OM, Pereira AM, Romijn JA. Treatment and follow-up of clinically nonfunctioning pituitary macroadenomas. J Clin Endocrinol Metab. 2008;93:3717–26.
Chen Y, Wang CD, Su ZP, Chen YX, Cai L, Zhuge QC, et al. Natural history of postoperative nonfunctioning pituitary adenomas: a systematic review and meta-analysis. Neuroendocrinology. 2012;96:333–42.
Mete O, Hayhurst C, Alahmadi H, Monsalves E, Gucer H, Gentili F, et al. The role of mediators of cell invasiveness, motility, and migration in the pathogenesis of silent corticotroph adenomas. Endocr Pathol. 2013;24:191–8.
Alahmadi H, Lee D, Wilson JR, Hayhurst C, Mete O, Gentili F, et al. Clinical features of silent corticotroph adenomas. Acta Neurochir. 2012;154:1493–8.
Lopez JA, Kleinschmidt-Demasters BB, Sze CI, Woodmansee WW, Lillehei KO. Silent corticotroph adenomas: further clinical and pathological observations. Hum Pathol. 2004;35:1137–47.
Scheithauer BW, Jaap AJ, Horvath E, Kovacs K, Lloyd RV, Meyer FB, et al. Clinically silent corticotroph tumors of the pituitary gland. Neurosurgery. 2000;47:723–9.
Drummond J, Roncaroli F, Grossman AB, Korbonits M. Clinical and pathological aspects of silent pituitary adenomas. J Clin Endocrinol Metab. 2019;104:2473–89.
Horvath E, Kovacs K, Smyth HS, Cusimano M, Singer W. Silent adenoma subtype 3 of the pituitary-immunohistochemical and ultrastructural classification: a review of 29 cases. Ultrastruct Pathol. 2005;29:511–24.
Erickson D, Scheithauer B, Atkinson J, Horvath E, Kovacs K, Lloyd RV, et al. Silent subtype 3 pituitary adenoma: a clinicopathologic analysis of the Mayo Clinic experience. Clin Endocrinol. 2009;71:92–9.
George DH, Scheithauer BW, Kovacs K, Horvath E, Young WF Jr., Lloyd RV, et al. Crooke’s cell adenoma of the pituitary: an aggressive variant of corticotroph adenoma. Am J Surg Pathol. 2003;27:1330–6.
Di Ieva A, Davidson JM, Syro LV, Rotondo F, Montoya JF, Horvath E, et al. Crooke’s cell tumors of the pituitary. Neurosurgery. 2015;76:616–22.
Cortez GM, Monteiro A, Agnoletto G, Bit-Ivan EN, Sauvageau E, Hanel RA. Aggressive pituitary tumor with Crooke’s cells and invasion of the posterior fossa. World Neurosurg. 2020;138:530–4.
Bakhtiar Y, Hirano H, Arita K, Yunoue S, Fujio S, Tominaga A, et al. Relationship between cytokeratin staining patterns and clinico-pathological features in somatotropinomae. Eur J Endocrinol. 2010;163:531–9.
Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol. 2013;168:491–9.
Kiseljak-Vassiliades K, Carlson NE, Borges MT, Kleinschmidt-DeMasters BK, Lillehei KO, Kerr JM, et al. Growth hormone tumor histological subtypes predict response to surgical and medical therapy. Endocrine. 2015;49:231–41.
Horvath E, Kovacs K, Singer W, Smyth HS, Killinger DW, Ezrin C, et al. Acidophil stem cell adenoma of the human pituitary: clinicopathologic analysis of 15 cases. Cancer. 1981;47:761–71.
Horvath E, Kovacs K, Singer W, Ezrin C, Kerenyi NA. Acidophil stem cell adenoma of the human pituitary. Arch Pathol Lab Med. 1977;101:594–9.
Huang C, Ezzat S, Asa SL, Hamilton J. Dopaminergic resistant prolactinomas in the peripubertal population. J Pediatr Endocrinol Metab. 2006;19:951–3.
Brucker-Davis F, Oldfield EH, Skarulis MC, Doppman JL, Weintraub BD. Thyrotropin-secreting pituitary tumors: diagnostic criteria, thyroid hormone sensitivity, and treatment outcome in 25 patients followed at the National Institutes of Health. J Clin Endocrinol Metab. 1999;84:476–86.
Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G, et al. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol. 2013;126:123–35.
Di Ieva A, Rotondo F, Syro LV, Cusimano MD, Kovacs K. Aggressive pituitary adenomas-diagnosis and emerging treatments. Nat Rev Endocrinol. 2014;10:423–35.
Monsalves E, Juraschka K, Tateno T, Agnihotri S, Asa SL, Ezzat S, et al. The PI3K/AKT/mTOR pathway in the pathophysiology and treatment of pituitary adenomas. Endocr Relat Cancer. 2014;21:R331–44.
Donovan LE, Arnal AV, Wang SH, Odia Y. Widely metastatic atypical pituitary adenoma with mTOR pathway STK11(F298L) mutation treated with everolimus therapy. CNS Oncol. 2016;5:203–9.
Syro LV, Ortiz LD, Scheithauer BW, Lloyd R, Lau Q, Gonzalez R, et al. Treatment of pituitary neoplasms with temozolomide: a review. Cancer. 2011;117:454–62.
McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol. 2018;178:265–76.
Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, et al. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. 2018;178:G1–24.
Burman P, Lamb L, McCormack A. Temozolomide therapy for aggressive pituitary tumours - current understanding and future perspectives. Rev Endocr Metab Disord. 2020;21:263–76.
Bengtsson D, Schroder HD, Andersen M, Maiter D, Berinder K, Feldt RU, et al. Long-term outcome and MGMT as a predictive marker in 24 patients with atypical pituitary adenomas and pituitary carcinomas given treatment with temozolomide. J Clin Endocrinol Metab. 2015;100:1689–98.
Bush ZM, Longtine JA, Cunningham T, Schiff D, Jane JA Jr., Vance ML, et al. Temozolomide treatment for aggressive pituitary tumors: correlation of clinical outcome with O(6)-methylguanine methyltransferase (MGMT) promoter methylation and expression. J Clin Endocrinol Metab. 2010;95:E280–90.
Thearle MS, Freda PU, Bruce JN, Isaacson SR, Lee Y, Fine RL. Temozolomide (Temodar(R)) and capecitabine (Xeloda(R)) treatment of an aggressive corticotroph pituitary tumor. Pituitary. 2011;14:418–24.
Zacharia BE, Gulati AP, Bruce JN, Carminucci AS, Wardlaw SL, Siegelin M, et al. High response rates and prolonged survival in patients with corticotroph pituitary tumors and refractory Cushing disease from capecitabine and temozolomide (CAPTEM): a case series. Neurosurgery. 2014;74:E447–55.
Alshaikh OM, Asa SL, Mete O, Ezzat S. An institutional experience of tumor progression to pituitary carcinoma in a 15-year cohort of 1055 consecutive pituitary neuroendocrine tumors. Endocr Pathol. 2019;30:118–27.
Santos-Pinheiro F, Penas-Prado M, Kamiya-Matsuoka C, Waguespack SG, Mahajan A, Brown PD, et al. Treatment and long-term outcomes in pituitary carcinoma: a cohort study. Eur J Endocrinol. 2019;181:397–407.
Baldari S, Ferrau F, Alafaci C, Herberg A, Granata F, Militano V, et al. First demonstration of the effectiveness of peptide receptor radionuclide therapy (PRRT) with 111In-DTPA-octreotide in a giant PRL-secreting pituitary adenoma resistant to conventional treatment. Pituitary. 2012;15:S57–60.
Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med. 1954;16:363–71.
DeLellis RA. Multiple endocrine neoplasia syndromes revisited. Clinical, morphologic and molecular features. Lab Invest. 1995;72:494–505.
Scheithauer BW, Laws ER Jr., Kovacs K, Horvath E, Randall RV, Carney JA. Pituitary adenomas of the multiple endocrine neoplasia type I syndrome. Semin Diagn Pathol. 1987;4:205–11.
Trouillas J, Labat-Moleur F, Sturm N, Kujas M, Heymann MF, Figarella-Branger D, et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol. 2008;32:534–43.
Shintani Y, Yoshimoto K, Horie H, Sano T, Kanesaki Y, Hosoi E, et al. Two different pituitary adenomas in a patient with multiple endocrine neoplasia type 1 associated with growth hormone-releasing hormone-producing pancreatic tumor: clinical and genetic features. Endocr J. 1995;42:331–40.
Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. 2014;386:2–15.
Brennan P. Breast cancer risk in MEN1—a cancer genetics perspective. Clin Endocrinol. 2015;82:327–229.
Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103:15558–63.
Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. 2007;92:3321–5.
Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neop(2009)lasia type 1 and related states. J Clin Endocrinol Metab. 2009;94:1826–34.
Georgitsi M. MEN-4 and other multiple endocrine neoplasias due to cyclin-dependent kinase inhibitors (p27(Kip1) and p18(INK4C)) mutations. Best Pr Res Clin Endocrinol Metab. 2010;24:425–37.
Alrezk R, Hannah-Shmouni F, Stratakis CA. MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer. 2017;24:T195–208.
Nachtigall LB, Guarda FJ, Lines KE, Ghajar A, Dichtel L, Mumbach G, et al. Clinical MEN-1 among a large cohort of patients with acromegaly. J Clin Endocrinol Metab. 2020;105:e2271.
Lines KE, Nachtigall LB, Dichtel LE, Cranston T, Boon H, Zhang X, et al. Multiple endocrine neoplasia type 1 (MEN1) phenocopy due to a cell cycle division 73 (CDC73) variant. J Endocr Soc. 2020;4:bvaa142.
Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine. 1985;64:270–83.
Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92.
Yin Z, Williams-Simons L, Parlow AF, Asa S, Kirschner LS. Pituitary-specific knockout of the Carney complex gene prkar1a leads to pituitary tumorigenesis. Mol Endocrinol. 2008;22:380–7.
Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72:106–16.
Xekouki P, Stratakis CA. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer. 2012;19:C33–40.
Karamurzin Y, Zeng Z, Stadler ZK, Zhang L, Ouansafi I, Al Ahmadie HA, et al. Unusual DNA mismatch repair-deficient tumors in Lynch syndrome: a report of new cases and review of the literature. Hum Pathol. 2012;43:1677–87.
Bengtsson D, Joost P, Aravidis C, Askmalm SM, Backman AS, Melin B, et al. Corticotroph pituitary carcinoma in a patient with lynch syndrome (LS) and pituitary tumors in a nationwide LS cohort. J Clin Endocrinol Metab. 2017;102:3928–32.
Soares BS, Frohman LA. Isolated familial somatotropinoma. Pituitary. 2004;7:95–101.
Beckers A, Daly AF. The clinical, pathological, and genetic features of familial isolated pituitary adenomas. Eur J Endocrinol. 2007;157:371–82.
Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006;312:1228–30.
Georgitsi M, De Menis E, Cannavo S, Makinen MJ, Tuppurainen K, Pauletto P, et al. Aryl hydrocarbon receptor interacting protein (AIP) gene mutation analysis in children and adolescents with sporadic pituitary adenomas. Clin Endocrinol. 2008;69:621–7.
Pesatori AC, Baccarelli A, Consonni D, Lania A, Beck-Peccoz P, Bertazzi PA, et al. Aryl hydrocarbon receptor-interacting protein and pituitary adenomas: a population-based study on subjects exposed to dioxin after the Seveso, Italy, accident. Eur J Endocrinol. 2008;159:699–703.
Hernandez-Ramirez LC, Gabrovska P, Denes J, Stals K, Trivellin G, Tilley D, et al. Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J Clin Endocrinol Metab. 2015;100:E1242–54.
Trivellin G, Daly AF, Faucz FR, Yuan B, Rostomyan L, Larco DO, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014;371:2363–74.
Moran A, Asa SL, Kovacs K, Horvath E, Singer W, Sagman U, et al. Gigantism due to pituitary mammosomatotroph hyperplasia. N Engl J Med. 1990;323:322–7.
Asa SL, Mete O, Ezzat S. Genomics and epigenomics of pituitary tumors: what do pathologists need to know? Endocr Pathol. 2021;32:3–16.
Spada A, Arosio M, Bochicchio D, Bazzoni N, Vallar L, Bassetti M, et al. Clinical, biochemical and morphological correlates in patients bearing growth hormone-secreting pituitary tumors with or without constitutively active adenylyl cyclase. J Clin Endocrinol Metab. 1990;71:1421–6.
Ballmann C, Thiel A, Korah HE, Reis AC, Saeger W, Stepanow S, et al. USP8 mutations in pituitary Cushing adenomas-targeted analysis by next-generation sequencing. J Endocr Soc. 2018;2:266–78.
Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet. 2015;47:31–8.
Casar-Borota O, Boldt HB, Engstrom BE, Andersen MS, Baussart B, Bengtsson D, et al. Corticotroph aggressive pituitary tumours and carcinomas frequently harbour ATRX mutations. J Clin Endocrinol Metab. in press. 2021.
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–203.
Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6140.
Newey PJ, Nesbit MA, Rimmer AJ, Head RA, Gorvin CM, Attar M, et al. Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 2013;98:E796–800.
Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ, et al. Landscape of genomic alterations in pituitary adenomas. Clin Cancer Res. 2017;23:1841–51.
Hage M, Viengchareun S, Brunet E, Villa C, Pineau D, Bouligand J, et al. Genomic alterations and complex subclonal architecture in sporadic GH-secreting pituitary adenomas. J Clin Endocrinol Metab. 2018;103:1929–39.
De Sousa SMC, Wang PPS, Santoreneos S, Shen A, Yates CJ, Babic M, et al. The genomic landscape of sporadic prolactinomas. Endocr Pathol. 2019;30:318–28.
Ben-Shlomo A, Deng N, Ding E, Yamamoto M, Mamelak A, Chesnokova V, et al. DNA damage and growth hormone hypersecretion in pituitary somatotroph adenomas. J Clin Invest. 2020;130:5738–55.
Karpathakis A, Dibra H, Pipinikas C, Feber A, Morris T, Francis J, et al. Prognostic impact of novel molecular subtypes of small intestinal neuroendocrine tumor. Clin Cancer Res. 2016;22:250–8.
Ezzat S, Cheng S, Asa SL. Epigenetics of pituitary tumors: pathogenetic and therapeutic implications. Mol Cell Endocrinol. 2018;469:70–6.
Tiemensma J, Kokshoorn NE, Biermasz NR, Keijser BJ, Wassenaar MJ, Middelkoop HA, et al. Subtle cognitive impairments in patients with long-term cure of Cushing’s disease. J Clin Endocrinol Metab. 2010;95:2699–714.
Tiemensma J, Biermasz NR, van der Mast RC, Wassenaar MJ, Middelkoop HA, Pereira AM, et al. Increased psychopathology and maladaptive personality traits, but normal cognitive functioning, in patients after long-term cure of acromegaly. J Clin Endocrinol Metab. 2010;95:E392–402.
Sievers C, Ising M, Pfister H, Dimopoulou C, Schneider HJ, Roemmler J, et al. Personality in patients with pituitary adenomas is characterized by increased anxiety-related traits: comparison of 70 acromegalic patients with patients with non-functioning pituitary adenomas and age- and gender-matched controls. Eur J Endocrinol. 2009;160:367–73.
Barocas DA, Mitchell R, Chang SS, Cookson MS. Impact of surgeon and hospital volume on outcomes of radical prostatectomy. Urol Oncol. 2010;28:243–50.
Jawaid W, Chan B, Jesudason EC. Subspecialization may improve an esophageal atresia service but has not addressed declining trainee experience. J Pediatr Surg. 2012;47:1363–8.
Abdulla AG, Ituarte PH, Wiggins R, Teisberg EO, Harari A, Yeh MW. Endocrine surgery as a model for value-based health care delivery. Surg Neurol Int. 2012;3:163.
Skotko BG, Davidson EJ, Weintraub GS. Contributions of a specialty clinic for children and adolescents with Down syndrome. Am J Med Genet A. 2013;161A:430–7.
Nambudiri VE, Sober AJ, Kimball AB. Specializing in accountability: strategies to prepare a subspecialty workforce for care delivery redesign. Acad Med. 2013;88:1900–3.
Middleton LP, Feeley TW, Albright HW, Walters R, Hamilton SH. Second-opinion pathologic review is a patient safety mechanism that helps reduce error and decrease waste. J Oncol Pr. 2014;10:275–80.
Ho J, Ahlers SM, Stratman C, Aridor O, Pantanowitz L, Fine JL, et al. Can digital pathology result in cost savings? A financial projection for digital pathology implementation at a large integrated health care organization. J Pathol Inf. 2014;5:33.
Grady AT, Sosa JA, Tanpitukpongse TP, Choudhury KR, Gupta RT, Hoang JK. Radiology reports for incidental thyroid nodules on CT and MRI: high variability across subspecialties. AJNR Am J Neuroradiol. 2015;36:397–402.
Plotti F, Capriglione S, Miranda A, Scaletta G, Aloisi A, Luvero D, et al. The impact of gynecologic oncology training in the management of cancer patients: is it really necessary? A prospective cohort study. Eur J Obstet Gynecol Reprod Biol. 2015;184:19–23.
Pathik B, De Pasquale CG, McGavigan AD, Sinhal A, Vaile J, Tideman PA, et al. Subspecialisation in cardiology care and outcome: should clinical services be redesigned, again? Intern Med J. 2016;46:158–66.
Maharaj MM, Hogan JA, Phan K, Mobbs RJ. The role of specialist units to provide focused care and complication avoidance following traumatic spinal cord injury: a systematic review. Eur Spine J. 2016;25:1813–20.
Ray KN, Ashcraft LE, Kahn JM, Mehrotra A, Miller E. Family perspectives on high-quality pediatric subspecialty referrals. Acad Pediatr. 2016;16:594–600.
Volynskaya Z, Chow H, Evans A, Wolff A, Lagmay-Traya C, Asa SL. Integrated pathology informatics enables high-quality personalized and precision medicine: digital pathology and beyond. Arch Pathol Lab Med. 2018;142:369–82.
Nose V, Ezzat S, Horvath E, Kovacs K, Laws ER, Lloyd R, et al. Protocol for the examination of specimens from patients with primary pituitary tumors. Arch Pathol Lab Med. 2011;135:640–6.
Frara S, Rodriguez-Carnero G, Formenti AM, Martinez-Olmos MA, Giustina A, Casanueva FF. Pituitary tumors centers of excellence. Endocrinol Metab Clin North Am. 2020;49:553–64.
Kloppel G, Rindi G, Anlauf M, Perren A, Komminoth P. Site-specific biology and pathology of gastroenteropancreatic neuroendocrine tumors. Virchows Arch. 2007;451:S9–27.
Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. The 2019 WHO classification of tumours of the digestive system. Histopathology. 2020;76:182–8.
Nilsson AH. The gut as the largest endocrine organ in the body. Ann Oncol. 2001;12:S63–8.
Bosman FT, de la Riviere AB, Giard RW, Verhofstad AA, Cramer-Knijnenburg G. Amine and peptide hormone production by lung carcinoid: a clinicopathological and immunocytochemical study. J Clin Pathol. 1984;37:931–6.
Vanoli A, La RS, Klersy C, Grillo F, Albarello L, Inzani F, et al. Four neuroendocrine tumor types and neuroendocrine carcinoma of the duodenum: analysis of 203 cases. Neuroendocrinology. 2017;104:112–25.
Sohn JH, Cho MY, Park Y, Kim H, Kim WH, Kim JM, et al. Prognostic significance of defining L-cell type on the biologic behavior of rectal neuroendocrine tumors in relation with pathological parameters. Cancer Res Treat. 2015;47:813–22.
Kim JY, Kim KS, Kim KJ, Park IJ, Lee JL, Myung SJ, et al. Non-L-cell immunophenotype and large tumor size in rectal neuroendocrine tumors are associated with aggressive clinical behavior and worse prognosis. Am J Surg Pathol. 2015;39:632–43.
Lee SH, Kim BC, Chang HJ, Sohn DK, Han KS, Hong CW, et al. Rectal neuroendocrine and L-cell tumors: diagnostic dilemma and therapeutic strategy. Am J Surg Pathol. 2013;37:1044–52.
Sano T, Asa SL, Kovacs K. Growth hormone-releasing hormone-producing tumors: clinical, biochemical, and morphological manifestations. Endocr Rev. 1988;9:357–73.
La Rosa S, Volante M, Uccella S, Maragliano R, Rapa I, Rotolo N, et al. ACTH-producing tumorlets and carcinoids of the lung: clinico-pathologic study of 63 cases and review of the literature. Virchows Arch. 2019;475:587–97.
Asa SL, Kovacs K, Vale W, Petrusz P, Vecsei P. Immunohistologic localization of corticotrophin-releasing hormone in human tumors. Am J Clin Pathol. 1987;87:327–33.
Uccella S, Blank A, Maragliano R, Sessa F, Perren A, La RS. Calcitonin-producing neuroendocrine neoplasms of the pancreas: clinicopathological study of 25 cases and review of the literature. Endocr Pathol. 2017;28:351–61.
Maragliano R, Vanoli A, Albarello L, Milione M, Basturk O, Klimstra DS, et al. ACTH-secreting pancreatic neoplasms associated with Cushing syndrome: clinicopathologic study of 11 cases and review of the literature. Am J Surg Pathol. 2015;39:374–82.
Uccella S, Asa SL, Mete O. Neuroendocrine neoplasms of unknown primary site. In: Asa SL, La Rosa S, Mete O, editors. The spectrum of neuroendocrine neoplasia. Cham, Switzerland: Springer; Chapter 16, 2020;357–88.
Akirov A, Larouche V, Alshehri S, Asa SL, Ezzat S. Treatment options for pancreatic neuroendocrine tumors. Cancers. 2019;11:828.
Zandee WT, Kamp K, van Adrichem RC, Feelders RA, de Herder WW. Effect of hormone secretory syndromes on neuroendocrine tumor prognosis. Endocr Relat Cancer. 2017;24:R261–74.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Consortia
Contributions
Conception and design: SLA, OM, ABG, and SE; writing and editing: SLA, OM, MDC, IEM, AP, SY, HN, OC-B, SU, SLR, ABG, and SE; figures SLA and IEM. All authors approved the content and submission.
Attendees of the 15th Meeting of the International Pituitary Pathology Club, Istanbul October 2019
Sofia Asioli16, Süheyla Uyar Bozkurt17, Nil Comunoglu18, Giulia Cossu19, Peter Earls20, Nuperi Gazioglu21, Richard A. Hickman22, Hidetoshi Ikeda23, Emilija Manojlovic-Gacic24, Mahmoud Messerer19, Buge Öz25, Sara Pakbaz2, Federico Roncaroli26, Wolfgang Saeger27, John Turchini28, Sema Yarman29
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the Attendees of the 15th meeting of the International Pituitary Pathology Club are listed below Author contributions.
Rights and permissions
About this article

Cite this article
Asa, S.L., Mete, O., Cusimano, M.D. et al. Pituitary neuroendocrine tumors: a model for neuroendocrine tumor classification. Mod Pathol 34, 1634–1650 (2021). https://doi.org/10.1038/s41379-021-00820-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-021-00820-y
This article is cited by
-
Molecular Classification of Gastrointestinal and Pancreatic Neuroendocrine Neoplasms: Are We Ready for That?
Endocrine Pathology (2024)
-
Heterogeneity of TPIT expression in ACTH-secreting extra-pituitary neuroendocrine tumors (NETs) supports the existence of different cellular programs in pancreatic and pulmonary NETs
Virchows Archiv (2023)
-
The Spectrum of Familial Pituitary Neuroendocrine Tumors
Endocrine Pathology (2023)
-
Post-operative surveillance for somatotroph, lactotroph and non-functional pituitary adenomas after curative resection: a systematic review
Pituitary (2023)
-
The Spectrum of Endocrine Pathology
Endocrine Pathology (2023)
