
Abstract
Ancillary testing during the initial workup of acute myeloid leukemia (AML) is largely performed using aspirated materials. We utilized multiplex immunofluorescence (MIF) imaging with digital image analysis to perform an in situ analysis of the microenvironment in NPM1-mutated AML using diagnostic bone marrow biopsy tissues (N = 17) and correlated these findings with diagnostic next-generation sequencing (NGS, N = 17), flow cytometry (FC, N = 14), and first remission (CR1) NPM1-specific molecular MRD (n = 16) data. The total CD3-positive T-cell percentages correlated positively between FC and MIF (r = 0.53, p = 0.05), but were significantly lower by MIF (1.62% vs. 3.4%, p = 0.009). The percentage of mutant NPM1-positive (NPM1c+) cells ranged from 9.7 to 90.8% (median 45.4%) and did not correlate with the NPM1 mutant allele fraction by NGS (p > 0.05). The percentage of CD34+/NPM1c+ cells ranged from 0 to 1.8% (median 0.07%). The percentage of NPM1c+ cells correlated inversely (34% vs. 62%, p = 0.03), while the percentages of CD3−/NPM1c− cells (64% vs. 35%, p = 0.03), and specifically CD3−/CD4−/NPM1c− cells (26% vs. 13%, p = 0.04), correlated positively with subsequent MRD. Discordances between MIF and FC/NGS data suggest that aspirate materials are likely an imperfect reflection of the core biopsy tissue. Furthermore, increased numbers of NPM1 wild-type cells within the microenvironment at diagnosis correlate with the subsequent presence of MRD.
Similar content being viewed by others

Introduction
Acute myeloid leukemia (AML) with mutated nucleophosmin 1 gene (NPM1) is recognized as a separate entity in the revised 2016 World Health Organization (WHO) classification of myeloid neoplasms [1]. Its presence in ~30% of all cases, and in 50–60% of those with normal cytogenetics, makes NPM1 one of the most commonly observed mutations in AML [1,2,3,4,5,6,7,8]. NPM1 mutation is generally associated with a favorable clinical outcome, however, FLT3-ITD co-mutation has been shown to diminish the favorable NPM1 effect, particularly in the presence of mutant DNMT3A [9, 10]. Interrogation for mutant NPM1 and other genetic markers of prognostic and therapeutic significance in NPM1-mutated AML, in other AML subtypes, and in myeloid neoplasms more broadly, is typically performed by either “bulk” sequencing or single gene analyses using aspirate marrow material or peripheral blood.
The importance of co-mutations in FLT3 and DNMT3A in determining the biology and prognosis of NPM1-mutated AML is well-established at this time; however, in recent years, evidence has emerged that NPM1-mutated/CD34-positive leukemic stem cells [11, 12], as well as anti-leukemic T-cells stimulated by the MHC molecule-mediated presentation of mutant NPM1 protein may also play roles in the disease biology and prognosis [13,14,15]. Although the blast population in NPM1-mutated AML is classically found to be weakly-positive or negative for the stem cell marker CD34 at the time of diagnosis, independent studies published in 2010 and led by Martelli and Taussig, respectively, interrogated for the presence and leukemogenic potential of an NPM1-mutated/CD34-positive population [11, 12]. Martelli and colleagues identified NPM1 mutations in CD34-positive stem cells in each of the seventeen tested samples of NPM1-mutated AML. Transplantation of these cells into immunocompromised mice produced a CD34-negative leukemia that was morphologically and immunophenotypically similar to human NPM1-mutated AML [11]. Conversely, in another mouse model, work by Taussig and colleagues suggested that the leukemia-initiating clone (LIC) in NPM1-mutated AML may reside largely in the CD34-negative cell fraction.
In 2012, Greiner and colleagues first demonstrated by using primary AML samples and in vitro ELISPOT assays that mutant NPM1 protein is capable of eliciting both CD4-positive and CD8-positive T-cells responses. This initial finding raised the possibility that mutant NPM1 protein could be a promising target for immunotherapies in this AML subtype [13]. Subsequent studies have further demonstrated that mutant NPM1 protein-derived peptides can indeed be found in the HLA Class I ligandome of affected patients, that some patients may innately possess circulating anti-mutant-NPM1 T-cell clones, and that these T-cells are capable of lysing leukemic cells [14, 15].
It is important to note that the aforementioned studies have largely been performed using aspirated marrow and/or peripheral blood samples. Robust, quantitative, in situ analyses using human bone marrow biopsy tissues to identify CD34-positive LICs, anti-leukemic T-cell infiltrates, and other populations of potential prognostic and/or therapeutic interest in the microenvironment have as yet not been performed, and the marrow topography has not been formally evaluated. In this study, we utilized multiplexed immunofluorescence (MIF) staining, coupled with digital image analysis, to perform an exploration of the marrow tissue microenvironment in NPM1-mutated AML.
Methods
Tissue samples
Bouin-fixed, lightly decalcified (RapidCal-Immuno for 15 min), paraffin-embedded (BFPE) bone marrow core biopsy specimens from newly-diagnosed, treatment-naive NPM1-mutated AML patients were derived from the archives of Brigham and Women’s Hospital, Boston, MA, with institutional review board approval (IRB# 2009P001369). The original stained tissue sections and diagnostic reports were reviewed by two board-certified hematopathologists (S.S.P and O.K.W.). Seventeen cases were selected for the study, based on the availability of high-quality biopsy tissue of sufficient quantity for further evaluation, and particulate and cellular aspirate smear preparations. Inclusion criteria for biopsy quality included length >1.5 cm, and absence of architectural distortion, crush, or aspiration artifacts (on morphologic review of H&E-stained sections).
Immunohistochemical (IHC) and multiplexed immunofluorescence (MIF) studies
Immunohistochemistry to interrogate for mutant NPM1 protein (ThermoFisher Scientific, Rockford, IL; Catalog #: PA1-46356) was performed on 4-μm-thick BFPE sections from all diagnostic bone marrow biopsy samples using previously-established protocols [16]. Visual scoring to determine the percentage of positively-staining cells in each case was performed independently by two board-certified hematopathologists (S.S.P. and O.K.W.) and results were averaged.
Multiplexed immunofluorescence (mIF) was performed by staining 4-μm-thick BFPE bone marrow core biopsy sections in a BondRX automated stainer, using published protocols [17]. One panel of primary antibody/fluorophore pairs was applied to all cases (listed in order of application sequence): (1) anti-CD3 (A0452, 1:750, Dako), (2) anti-CD8 (C8/144B, 1:7000, Abcam), (3) anti-CD4 (4B12, 1:250, Invitrogen), (4) anti-CD34 (QBEnd/10, 1:10,500, BioLegend), (5) anti-Granzyme B (GrB-7, 1:100, Invitrogen), and (6) anti-mutant NPM1-specific antibody (as above, 1:2500). Antibody/Opal fluor combinations were utilized as follows: CD3/650 (1:100), CD8/540 (1:100), CD4/520 (1:100), CD34/570 (1:200), Granzyme B/620 (1:200), and anti-mutant NPM1/690 (1:50). All slides were also stained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear identification.
Image acquisition
For each slide stained by MIF, three to six representative regions of interest (based on the availability of evaluable marrow) were captured at ×20 resolution as multispectral images using the Mantra Quantitative Pathology Workstation (PerkinElmer [now Akoya Biosciences], Hopkinton, MA).
Cell identification
After image capture, the fields of view were spectrally unmixed and analyzed using supervised machine learning algorithms within Inform 2.4.2 (Akoya), which assigns phenotypes to all cells in the image, according to a combination of IF characteristics associated with segmented nuclei (DAPI). Each cell phenotype-specific algorithm is based on an iterative training/test process, whereby a small number of cells (training phase, typically 15–20 cells) are manually selected by the pathologist as being most representative of each phenotype of interest and the algorithm then predicts the phenotype for all remaining cells (testing phase) [18]. The decisions made by the software can be overruled to improve accuracy until phenotyping is optimized. Unique phenotyping was performed for each biopsy sample, and then applied to individually-captured study images (Mantra). Thresholds for “positive” staining and the accuracy of phenotypic algorithms were confirmed by the primary study pathologist (S.S.P.) for each case. InForm (Akoya) automatically derives maps of cell membranes and Cartesian coordinates for each phenotyped cell within the image.
Quantification of the microenvironment
Quantities, spatial attributes, and graphical abstractions were generated from InForm-exported images and metadata by our image analysis pipeline, which extends our previously reported methods [17], and is available at https://github.com/jason-weirather/pythologist. The percentages of each cell phenotype were measured on a per-case basis (e.g., percent mutant NPM1 [NPM1c]-positive, CD3-positive, etc.).
Hypothesis testing was performed using two-tailed non-parametric testing. Differences in quantities of cell phenotype-count densities or cell-phenotype proportions between MRD-positive and MRD-negative patients was tested using a two-tailed Mann–Whitney U test. Correlations between quantities generated by FC or NGS methodologies and MIF were performed by the Spearman test; the Wilcoxon test was used to evaluate for differences between paired samples. All data outputs are provided parenthetically as median values (across analyzed cases) followed by full ranges, unless otherwise stated. Statistical significance was set at p < 0.05, where appropriate.
Flow cytometric (FC) immunophenotyping
Data from flow cytometric analysis performed at BWH on aspirated materials obtained concurrently with the core biopsy samples were retrieved for a subset of cases (n = 14). Testing was performed on a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA), with data analyzed using FACSDiva software (BD Biosciences). Immunophenotypic data was retrieved for expression of the following markers for all cases: CD45, CD3, and CD34.
Next-generation sequencing studies (NGS)
Targeted NGS studies were performed on aspirated marrow materials obtained concurrently with the core biopsy samples at diagnosis in all cases (n = 17) using a previously-described myeloid mutation panel [19, 20]. The mutant NPM1 allele fraction (variant allele fraction, VAF) was recorded for all cases. MIF data outputs were additionally correlated with NPM1-specific MRD data obtained at first remission in a subset of case (n = 16), as previously reported [16]. In brief, MRD was measured with an NPM1-specific deep sequencing assay using bone marrow material obtained at the first remission timepoint.
Results
Patient Characteristics
This study included 17 de novo AML patients with mutated NPM1 (M:F = 0.5) for whom high quality diagnostic bone marrow biopsy tissue was available (see “Methods” section). The median age at diagnosis was 58 years (range: 27–72). The median WBC and bone marrow blast counts at diagnosis were 33 K/uL (range: 1.17–129.6 K/uL) and 65% (range: 26–89%), respectively. All patients were treated with one cycle of standard induction chemotherapy. MRD evaluation was also performed previously on marrow samples obtained at the time of first remission (CR1) in 16 patients; eight of these patients were previously deemed MRD-positive, and eight were MRD-negative by this testing [16].
Visual validation of MIF staining and cellular quantification
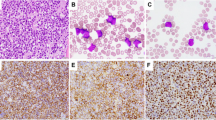
Visual inspection of all cases stained by MIF revealed appropriate and evaluable staining for all of the principally-evaluated markers (Fig. 1, representative case). The total nucleated cell count obtained ranged from 6914 to 23,041 across the 17 analyzed cases (median 13,176) (Fig. 2a). IHC for mutant NPM1 protein was performed in all 17 biopsy samples, and visually (median 90%, range 40–95%) correlated with the fraction of NPM1c+ cells as output by MIF analysis (median 45.4%, range 9.7–90.8%,) (Fig. 2b, representative cases shown). There was a significant positive correlation between visual scoring of IHC-stained slides and paired MIF outputs (r = 0.6, p = 0.02).

A representative case is shown (NPM1–6). Top: DAPI alone (left), DAPI/NPM1c (middle), DAPI/NPM1c/CD34 (right). Bottom: DAPI/CD3 (left), DAPI/NPM1c/CD4 (middle), DAPI/NPM1c/CD8 (right).

a Representative case of NPM1-mutated AML (NPM1–6) prepared by MIF. DAPI alone (top left) and paired phenotype map (top right). The following phenotypes are shown: NPM1c+CD34−CD4− (yellow), NPM1c+CD34−CD4+ (purple), NPM1c+CD34+CD4− (orange), NPM1c+CD34+CD4+ (pink), Other+CD34−CD4+ (green), Other+CD34+CD4− (brown), CD3+C8+CD4− (red), CD3+CD8−CD4+ (blue). Total evaluated nucleated cells ranged from 6914 to 23,041 across cases (median 13,176 cells). b Top: The NPM1c+ fraction of total cells ranged from 9.7 to 90.8% across analyzed cases (median 45.4%) [left]. Paired field of view from one representative case (NPM1–6) show IHC (left) and MIF (right) staining [right]. Bottom: Paired immunohistochemical staining for mutant NPM1 protein performed in representative cases at either end of the range for the cohort (NPM1–30, high expression, left; NPM1–62, low expression, right) are shown.
Quantification of T-cells by FC and MIF
By FC performed on 14 marrow aspirate samples at the time of diagnosis, the percentage of CD3-positive T-cells ranged from 0.6 to 24.5% (median 3.4%). By MIF performed on the 17 concurrently-obtained bone marrow core biopsy samples, total CD3-positive T-cells were defined by summing the cell counts for the following phenotypes: CD3+CD4+CD8+, CD3+CD4+CD8−, CD3+CD4−CD8+, and CD3+CD4−CD8−. The resulting percentage of CD3-positive T-cells ranged from 0.4 to 6.9% (median 1.9%, n = 17). Comparison of FC and MIF methodologies for total T-cell quantification revealed a borderline-significant positive correlation (r = 0.53, p = 0.05, N = 14); the percentage of T-cells measured by MIF was significantly lower than the percentage of T-cells measured by FC (1.62% vs. 3.4%, p = 0.009, N = 14) (Fig. 3). There was no difference in total CD3-positive T-cell percentage measured by MIF with respect to the presence or absence of any common co-mutations (those identified in >25% of the cohort, including DNMT3A, IDH1, FLT3-TKD, and FLT3-ITD).

a There is a borderline-significant positive correlation between methodologies for T-cell quantification (r = 0.53, p = 0.05, N = 14 pairs). The quantification of total T-cells is lower by MIF than by FC across cases (median 1.62% vs. 3.4%, p = 0.009, N = 14 pairs, Wilcoxon matched pairs test).
Quantification of CD34+ and/or NPM1-mutated (NPM1c+) cells
By FC performed on aspirated marrow samples, the percentage of cells within the dim-CD45-positive blast gate ranged from 21.8 to 90.4% (median 67.3%, N = 14). The CD34-positive fraction of cells in the blast gate ranged from 0 to 99.5% (median 0.5%, N = 14), and as a fraction of total cells ranged from 0 to 87.4% (median 0.2%, N = 14). By MIF, total CD34-positive cells ranged from 0.01 to 10% (median 0.42%, N = 17). Comparison of FC and MIF methodologies for total CD34-positive cell quantification revealed a borderline-significant positive correlation between them (r = 0.54, p = 0.05, N = 14), and no significant difference between the median output values between methods (p > 0.05). The percentage of mutant NPM1-positive (NPM1c+) cells ranged from 9.7 to 90.8% (median 45.4%) (Fig. 2b) and did not correlate with the NPM1 mutant allele fraction (p > 0.05) as measured by NGS. The percentage of CD34+NPM1c+ cells ranged from 0 to 1.8% (median 0.07%) (Fig. 4, representative case shown). There was no significant difference in the percentage of NPM1c+ cells with respect to gender or patient age greater than or less than 60 years. There was no significant correlation between the NPM1c+ percentage and the WBC or platelet counts or the marrow blast percentage as determined by either flow cytometry (dim-CD45 blast gate) or morphologic evaluation of the aspirate smear at the time of diagnosis (p > 0.05). The percentage of NPM1c+ cells was higher in IDH1 co-mutated (N = 7) relative to IDH1-wild type cases (N = 10) [59% vs. 34%, p = 0.04]; however, there was no difference in the NPM1c+ percentage with respect to the presence or absence of co-mutations in DNMT3A, FLT3-TKD, or FLT3-ITD.

Scattered CD34-positive cells are seen in a representative case of NPM1-mutated AML analyzed by MIF (NPM1–6, top left). A small subset of CD34-positive cells are also NPM1c+ (white arrows, bottom left) while most are NPM1c-negative (yellow arrows, bottom left). The CD34+/NPM1c+ fraction of total cells ranged from 0 to 1.8% across analyzed cases (median 0.07%).
Cell Phenotype Correlations with MRD at first remission
We further explored the relationship between various cell populations identified by MIF and the presence or absence of MRD at first remission. There was no difference between MRD-positive (n = 8) and MRD-negative (n = 8) cases with respect to the percentages of CD3+ T-cells or CD34+NPM1c+ cells (p > 0.05). We additionally identified no difference in the ratios of CD3+:NPMc+ cells between MRD-positive and MRD-negative samples (medians of per case values: 0.040 vs. 0.044, p > 0.05), or the ratios of CD3+:CD34 + NPM1c+ cells between MRD+ (n = 8) and MRD− (n = 7) samples where ≥1 CD34+NPM1c+ cell was identified (medians of per case values: 42.4 vs. 33.1, p > 0.05). Interestingly, the percentage of NPM1c+ cells was lower in MRD-positive versus MRD-negative cases (34% vs. 62%, p = 0.03), while the percentages of CD3-NPM1c- cells (64% vs. 35%, p = 0.03), and specifically the CD3−CD4–NPM1c– cells (26% vs. 13%, p = 0.04), were higher in MRD-positive versus MRD-negative cases (Fig. 5).

a The proportion of NPM1c+ cells correlates inversely (34% vs. 62%, p = 0.03) with the presence of MRD. b The proportion of CD3-NPM1c- cells (64% vs. 35%, p = 0.03) correlates positively with the presence of MRD. c The CD3−CD4−NPM1c− cells (26% vs. 13%, p = 0.04) correlates positively with the presence of MRD.
Discussion
Risk stratification of NPM1-mutated AML and other leukemia subtypes is largely based on ancillary testing performed on peripheral blood and/or aspirated bone marrow material. These tests include conventional morphologic examination of smear preparations, flow cytometric immunophenotyping, and targeted single-gene or panel-based polymerase chain reaction-based sequencing. Although aspirated materials are typically presumed to accurately represent the core biopsy tissue itself, hemodilution and/or any disproportionate sampling of the various marrow constituents may adversely affect the results of downstream ancillary testing and potentially impact subsequent patient management. In this study, we utilized multiplexed immunofluorescence (MIF) staining with digital image analysis to perform a quantitative in situ analysis of specific marrow cell types and correlated these data with the results of orthogonal techniques, including flow cytometry (FC) and next-generation sequencing (NGS) performed concurrently on aspirated materials. Moreover, we specifically interrogated for CD34-positive/NPM1-mutated cells, and mature T-cells, given recent reports of their potential significance in this leukemia subtype.
Given the relative novelty of this approach for use in decalcified, BFPE samples, we first aimed to ensure reliable staining and image preparation (Fig. 1). It is important to note that while conventional flow cytometric immunophenotyping typically evaluates at least 50,000-200,000 cells/events (depending on the laboratory and sample type), the number of cells evaluated by MIF in bone marrow biopsy samples is more limited. However, this limitation is offset by the ability of MIF to enable visualization of concurrent surface membrane, cytoplasmic, and nuclear protein expression on individual cell types, and to visualize and quantify the spatial relationships between these cells.
We found that quantification of specific cell types or leukemic disease burden by MIF was not completely concordant with FC and NGS testing. We observed a weak positive correlation between MIF and FC for quantification of mature T-cells, although outputs were statistically significantly higher by FC than by MIF. Of note, the anti-CD3 clone utilized for MIF analyses targets the epsilon chain of the protein, which can also be present in natural killer (NK) cells. We, therefore, suspect that the true discrepancy between mature T-cell quantitation by MIF and FC may be slightly greater than reflected by our analyses. Similarly, we did not observe a significant correlation between the fraction of NPM1-mutated cells (NPM1c+ cells) as assessed by MIF and the mutant NPM1 variant allele fraction by NGS. Together, these findings suggest that aspirated materials concurrently obtained at the time of the bone marrow biopsy procedure are a variable and potentially imperfect representation of the marrow tissue contents. The possibility of hemodilution affecting aspirate materials must be considered; however, we hypothesize that certain poorly-aspirated cell types, including stromal cells, adipocytes, and/or endothelial cells, by virtue of being underrepresented in aspirated material, may result in artificial enrichment of other marrow populations (e.g., leukemic and immune cells). This possibility is supported at least in part by the observed higher proportion of T-cells quantified by FC relative to MIF.
It has been previously demonstrated in NPM1-mutated AML that the NPM1 mutation, at least in some cases, can be ubiquitous within the myeloid compartment [21,22,23]. However, morphologic quantification of blast cells specifically remains a mainstay of diagnosis and the evaluation of therapy response. The potential significance of quantifying total cellular mutant NPM1 protein, as a reflection of the overall disease burden, has not yet been formally evaluated. In this initial exploration, we find that the fraction of NPM1c+ cells in the marrow as quantified by MIF fails to correlate with the marrow blast count as assessed by either morphology or FC. This finding serves to illustrate the potential discrepancy between the perceived burden of disease as assessed by the conventional morphologic measurement of blast percentage, and the true disease burden by quantitation of mutant protein expression.
Given the importance of MRD in the prognostication of NPM1-mutated AML, we attempted to identify any potential relationships between cell types present in the marrow tissue at diagnosis and the subsequent presence or absence of MRD. Because CD34-positive/NPM1-mutated LICs have been previously shown to incite NPM1-mutated AML in immunocompromised mice [11], we hypothesized that these LICs might be more resistant to conventional chemotherapy and/or provide a nidus for recurrent disease, and therefore that patients with identifiable, or larger populations of, CD34+NPM1c+ cells at diagnosis would be more likely to be MRD-positive at first remission. However, at least in this cohort, we did not find such a relationship. Likewise, we identified no correlation between the population size of mature T-cells at diagnosis as determined by MIF and the subsequent presence of MRD. Interestingly, however, we found that the total percentage of NPM1c+ cells at diagnosis was lower in patients who were subsequently found to be MRD-positive, while the percentages of CD3−NPM1c− cells, and specifically the CD3−CD4−NPM1c− cells, were higher (Fig. 5). We are limited in this initial study in our ability to further characterize these NPM1-wild type cells but hypothesize that they may represent some combination of stromal and/or endothelial cells, as these populations have been shown in certain solid tumors to support the neoplastic phenotype [24,25,26]. Using CD4 as a surrogate marker for cells of the monocyte/macrophage lineage, we might conclude that NPM1 wild-type monocytes/macrophages may not be as critical to NPM1-mutant cell survival as tumor-associated macrophages (TAMs) reported in other neoplastic contexts; [27,28,29,30] however, other cell types, including NPM1 wild-type erythroid precursors, megakaryocytes, osteoblasts, osteoclasts, B-cells, plasma cells, and/or NK cells, warrant further investigation to more precisely determine their biologic relevance in this specific disease [31,32,33].
The observed inverse relationship between the percentage of NPM1-mutated cells at diagnosis and the subsequent presence of MRD is somewhat unexpected, particularly in light of our recent findings which have established links between higher diagnostic NPM1 VAF, subsequent MRD, and inferior patient outcome [16, 20]. Given the lack of a correlation between the percentage of NPM1c+ cells and NPM1 VAF identified in this study, we hypothesize that VAF values may also be affected by disproportionate sampling of the marrow contents. We note that the diagnostic VAF was higher in MRD-positive versus MRD-negative cases, in this limited cohort (0.41 vs. 0.37, p > 0.05). It is conceivable that both diagnostic VAF and the percentage of NPMc+ cells within the biopsy tissue are relevant measurements for understanding the underlying biology and for prognostication of this leukemia subtype.
In summary, we have found in this initial study using multiparametric in situ analysis that the quantitation of specific bone marrow populations in NPM1-AML by MIF in the bone marrow biopsy and by orthogonal methodologies (FC and NGS) performed on bone marrow aspirate material, is not absolutely concordant, suggesting that aspirate materials are likely an imperfect reflection of the core biopsy tissue. In situ analyses further raise the possibility that increased numbers of NPM1 wild-type cells within the microenvironment at diagnosis may be associated with, or promote, leukemia cell survival and be of potential prognostic significance. Additional higher-order multiparametric in situ analyses are currently being pursued, the findings from which will be necessary to better define the NPM1-wild type populations identified in this study.
References
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. IARC, Lyon.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–43.
Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl J Med. 2012;366:1079–89.
Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl J Med. 2013;368:2059–74.
Ostronoff F, Othus M, Lazenby M, Estey E, Appelbaum FR, Evans A, et al. Prognostic significance of NPM1 mutations in the absence of FLT3-internal tandem duplication in older patients with acute myeloid leukemia: a SWOG and UK National Cancer Research Institute/Medical Research Council report. J Clin Oncol. 2015;33:1157–64.
Röllig C, Bornhäuser M, Kramer M, Thiede C, Ho AD, Krämer A, et al. Allogeneic stem-cell transplantation in patients with NPM1-mutated acute myeloid leukemia: results from a prospective donor versus no-donor analysis of patients after upfront HLA typing within the SAL-AML 2003 trial. J Clin Oncol. 2015;33:403–10.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl J Med. 2005;352:254–66.
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107:4011–20.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl J Med. 2016;374:2209–21.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47.
Martelli MP, Pettirossi V, Thiede C, Bonifacio E, Mezzasoma F, Cecchini D, et al. CD34+ cells from AML with mutated NPM1 harbor cytoplasmic mutated nucleophosmin and generate leukemia in immunocompromised mice. Blood. 2010;116:3907–22.
Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood. 2010;115:1976–84.
Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Götz M, et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood. 2012;120:1282–9.
van der Lee DI, Reijmers RM, Honders MW, Hagedoorn RS, de Jong RC, Kester MG, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest. 2019;129:774–85.
Kuželová K, Brodská B, Schetelig J, Röllig C, Ráčil Z, Walz JS, et al. Association of HLA class I type with prevalence and outcome of patients with acute myeloid leukemia and mutated nucleophosmin. PLoS ONE. 2018;13:e0204290.
Patel SS, Pinkus GS, Ritterhouse LL, Segal JP, Dal Cin P, Restrepo T, et al. High NPM1 mutant allele burden at diagnosis correlates with minimal residual disease at first remission in de novo acute myeloid leukemia. Am J Hematol. 2019;94:921–8.
Carey CD, Gusenleitner D, Lipschitz M, Roemer MGM, Stack EC, Gjini E, et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood. 2017;130:2420–30.
Feng Z, Puri S, Moudgil T, Wood W, Hoyt CC, Wang C, et al. Multispectral imaging of formalin-fixed tissue predicts ability to generate tumor-infiltrating lymphocytes from melanoma. J Immunother Cancer. 2015;3:47.
Kluk MJ, Lindsley RC, Aster JC, Lindeman NI, Szeto D, Hall D, et al. Validation and implementation of a custom next-generation sequencing clinical assay for hematologic malignancies. J Mol Diagn. 2016;18:507–15.
Patel SS, Kuo FC, Gibson CJ, Steensma DP, Soiffer RJ, Alyea EP, et al. High NPM1-mutant allele burden at diagnosis predicts unfavorable outcomes in de novo AML. Blood. 2018;131:2816–25.
Falini B, Martelli MP, Bolli N, Bonasso R, Ghia E, Pallotta MT, et al. Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood. 2006;108:1999–2005.
Pasqualucci L, Liso A, Martelli MP, Bolli N, Pacini R, Tabarrini A, et al. Mutated nucleophosmin detects clonal multilineage involvement in acute myeloid leukemia: Impact on WHO classification. Blood. 2006;108:4146–55.
Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109:874–85.
Hida K, Maishi N, Annan DA, Hida Y. Contribution of tumor endothelial cells in cancer progression. Int J Mol Sci. 2018;19:E1272.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22.
Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54.
Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl J Med. 2010;362:875–85.
Al-Matary YS, Botezatu L, Opalka B, Hönes JM, Lams RF, Thivakaran A, et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a Growth factor independence 1 dependent manner. Haematologica. 2016;101:1216–27.
Yang M, McKay D, Pollard JW, Lewis CE. Diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018;78:5492–503.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416.
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506:240–4.
An G, Acharya C, Feng X, Wen K, Zhong M, Zhang L, et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood. 2016;128:1590–603.
Wouters MCA, Nelson BH. Prognostic significance of tumor-infiltrating b cells and plasma cells in human cancer. Clin Cancer Res. 2018;24:6125–35.
Author information
Authors and Affiliations
Contributions
S.S.P. designed the study, performed research, and wrote the paper. O.K.W. contributed to study design, data collection, and paper preparation. S.J.R. provided reagents. M.L. and G.S.P. performed research. J.L.W. analyzed data. O.P. and E.F.M collected data. R.P.H. and G.I. analyzed data and edited the paper. All authors reviewed and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Patel, S.S., Lipschitz, M., Pinkus, G.S. et al. Multiparametric in situ imaging of NPM1-mutated acute myeloid leukemia reveals prognostically-relevant features of the marrow microenvironment. Mod Pathol 33, 1380–1388 (2020). https://doi.org/10.1038/s41379-020-0498-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0498-z
This article is cited by
-
NPM1 Biology in Myeloid Neoplasia
Current Hematologic Malignancy Reports (2020)
