
Abstract
Pleomorphic LCIS (P-LCIS) and florid LCIS (F-LCIS) are morphologic variants distinguished from classic LCIS by marked nuclear pleomorphism and/or an expansile growth pattern with or without necrosis. Given the rarity of these LCIS variants, little data exist regarding their molecular pathogenesis, natural history, and optimal management. The purpose of this study was to genomically profile LCIS variants to gain further insight into their biology. Nineteen cases of pure LCIS variants (17 P-LCIS, 2 F-LCIS) diagnosed on core needle biopsy at our institution from 2006 to 2017 were included, five of which were upgraded to invasive cancer at excision. Macrodissected lesions were analyzed by a hybrid-capture next generation sequencing assay that surveyed exonic sequences of 447 genes for mutations and copy number variations (CNVs) and 191 regions across 60 genes for structural rearrangements. LCIS variants were all confirmed as E-cadherin negative by immunohistochemistry. Receptor profiles among the 17 P-LCIS cases included HR+/HER2− (nine cases), HR+/HER2+ (three cases), HR−/HER2+ (two cases), and HR−/HER2− (three cases). The two F-LCIS cases were HR+/HER2− and HR+/HER2+. All LCIS variants had genetic alterations consistent with a lobular phenotype including 1q gain (16 cases), 16q loss (18 cases), and CDH1 mutations (18 cases). Highly recurrent ERBB2 alterations were noted including mutations (13 cases) and amplifications (six cases). Other significant alterations included mutations in PIK3CA (six cases), RUNX1 (four cases), ERBB3 (four cases), and CBFB (three cases), as well as amplification of CCND1 (five cases). A TP53 mutation was identified in one case of HR−/HER2+ P-LCIS with signet ring cell features that lacked 1q gain and 16q loss. P-LCIS and F-LCIS contain genetic alterations characteristic of lobular neoplasia; however, these LCIS variants are distinguished from classical LCIS reported in the literature by their highly recurrent ERBB2 alterations.
Similar content being viewed by others

Introduction
Lobular carcinoma in situ (LCIS), a risk factor for and nonobligate precursor of invasive breast cancer, consists of a neoplastic proliferation of poorly cohesive cells that fill and distend the acini of the mammary terminal duct lobular units (TDLUs). As first described in detail by Foote and Stewart [1] and further characterized by Haagensen et al. [2], “classic” LCIS refers to a low-to-intermediate grade lesion encompassing two cytomorphologies: (1) small cells with scant cytoplasm and small, uniform, round-to-oval nuclei that lack nucleoli (Haagensen type A cells) and (2) larger cells with more abundant cytoplasm, slightly pleomorphic nuclei, and nucleoli (Haagensen type B cells). Classic LCIS (C-LCIS) is not associated with specific clinical, radiologic, or gross findings, but rather presents as an incidental microscopic finding, and is commonly multicentric or bilateral.
A hallmark of LCIS is the loss of expression and/or function of membranous E-cadherin, a transmembrane glycoprotein encoded by the CDH1 gene on the long arm of chromosome 16 (16q22.1). E-cadherin, as a component of the adherens junction, plays an integral role in intercellular adhesion, in part by linking to the actin cytoskeleton via α-, β-, γ-, and p120 catenins. Immunohistochemical staining for E-cadherin and other members of the cadherin–catenin complex can be employed to distinguish LCIS from ductal carcinoma in situ (DCIS), the lack of E-cadherin and β-catenin staining and aberrant cytoplasmic p120 staining standing in contrast to the intact membranous pattern seen in the latter [3, 4]. This is clinically important due to differences in the presentation, natural history, and management of these lesions. In contrast to DCIS, which is considered a nonobligate precursor of invasive cancer and managed by complete surgical excision with or without radiotherapy, C-LCIS has been managed more conservatively as a risk factor for invasive cancer with risk reduction strategies [5, 6].
With the advent of E-cadherin immunohistochemistry in the 1990s, there has been an appreciation of the heterogeneity of LCIS and recognition of morphologic variants that in the past were often erroneously classified as DCIS [7, 8]. At present, two uncommon variants of LCIS, pleomorphic LCIS (P-LCIS) and florid LCIS (F-LCIS), are recognized in the World Health Organization (WHO) classification. P-LCIS is characterized by large cells with marked nuclear pleomorphism (nuclei > 4 times size of a lymphocyte or equivalent to those seen in high-grade DCIS), often with apocrine or signet ring cell features. These lesions commonly display a florid growth pattern with massive distention of TDLUs and comedo necrosis. A second and lesser known variant, F-LCIS (also referred to in the past as “LCIS with necrosis”), also exhibits marked distention of TDLUs or ducts, creating a confluent mass-like architecture, but has the cytologic features of C-LCIS (type A and/or type B cells). Its diagnosis requires the presence of at least one of two architectural features: markedly distended acini of involved TDLUs with little to no intervening stroma and/or an expanded acinus or duct filling at least one high-power field (an area equivalent to ~40–50 cells in diameter). Comedo necrosis may be present in the involved spaces [9,10,11,12,13,14].
As compared with C-LCIS, which is almost always hormone receptor (HR)-positive and human epidermal growth factor receptor 2 (HER2)-negative, LCIS variants show greater variability in receptor expression, with estrogen receptor (ER)-negativity in up to 50% and HER2-positivity in up to 30% [15]. P-LCIS is more likely to present as a targeted radiologic lesion, either calcifications or rarely a mass-forming lesion, and is associated with significantly higher upgrade rates on surgical excision than C-LCIS (25–40% [16,17,18,19] versus <5% [20,21,22,23]). However, little is known regarding the natural history of these LCIS variants [12, 24,25,26,27] due to their rarity and the historical tendency to treat them as DCIS, leading to controversy regarding their management, in particular whether they should be managed similar to C-LCIS or DCIS.
A better understanding of the molecular pathogenesis of LCIS and its variants may lead to improvements in classification and inform clinical management. Until recently, next generation sequencing (NGS)-based approaches had not been applied to these lesions, with the repertoire of somatic mutations and gene-level amplifications and deletions remaining to be discovered. Given this, the aim of our study was to perform genomic profiling of the pleomorphic and florid variants of LCIS using our internally-developed targeted NGS-based platform (OncoPanel).
Methods
Case selection
Following institutional review board approval, 44 consecutive cases of LCIS variants diagnosed as the highest risk lesion on core needle biopsy at Brigham and Women’s Hospital and Brigham and Women’s Faulkner Hospital between 2006 and 2017 were identified by key word search. As terminology for these variants has evolved over the years at our institutions, the key word search was constructed to capture cases that had been diagnosed as LCIS with “variant”, “non-classical” or “pleomorphic” features or carcinoma in situ with “ductal and lobular features”. Thirty-five cases had both core needle biopsy and excision specimens available for pathologic review, with the lesion on biopsy meeting histologic criteria for P-LCIS or F-LCIS found in the current WHO classification [9, 11]. Core needle biopsy specimens were selected for genomic profiling as they appeared to preferentially contain discrete and relatively pure LCIS lesions that correlated with the radiologic target. Of the 35 cases, 23 were deemed to have sufficient lesion on the biopsy to be submitted for genomic profiling. Nineteen cases that were successfully profiled constituted the final study population, including 17 cases of P-LCIS and two cases of F-LCIS. Among the 17 cases of P-LCIS, five were upgraded to invasive carcinoma and one to DCIS on surgical excision. One of the two cases of F-LCIS was upgraded to DCIS. Of the 19 cases included in this study, 15 have been previously reported by our group as part of a clinicopathologic study evaluating the rate of upgrade to invasive breast cancer or DCIS following a core needle biopsy diagnosis of nonclassic LCIS [16].
Immunohistochemistry
Receptor studies and immunostains for components of the cadherin–catenin complex performed as part of the original clinical case were reviewed. For cases, in which these studies were unavailable, immunohistochemistry was completed on 4-µm-thick formalin-fixed paraffin-embedded whole sections using antibodies directed against ER (SP1 rabbit monoclonal antibody from Thermo Fisher Scientific, Waltham, MA), progesterone receptor (PgR 636 rabbit monoclonal antibody, DAKO, Santa Clara, CA), human epidermal growth factor receptor 2 (SP3 rabbit monoclonal antibody, Cell Marque, Rocklin, CA), E-cadherin (NCH-38, DAKO, Santa Clara, CA), p120 (98/PP120, BD Biosciences, San Jose, CA), and β-catenin (14, BD Biosciences, San Jose, CA). Antigen retrieval was performed with 1 mM EDTA (pH 8.0) in a pressure cooker. External controls stained appropriately.
Interpretation of HR and HER2 immunohistochemical stains was adapted from the most recent American Society of Clinical Oncology/American College of Pathologists guidelines for breast cancer [28,29,30]. ER and PR were scored as “positive” (nuclear reactivity in >10% of lesional cells), “low positive” (nuclear reactivity in ≥1% of lesional cells) or “negative” (<1%), whereas HER2 was scored as “positive” (strong, complete membranous staining in a contiguous focus comprising at least 10% of the lesion), “equivocal” (weak-to-moderate membranous staining in a focus comprising at least 10% of the lesion), or “negative” (lesser degrees of staining). A final HER2 status of “positive” was assigned to cases scored as positive and “negative” to cases scored as either negative or equivocal. Lesions were interpreted as having “intact” or “reduced-to-absent” membranous staining for E-cadherin and β-catenin and “intact membranous” or “aberrant cytoplasmic” staining for p120 catenin.
Targeted capture massively parallel sequencing
Samples were processed in the Center for Advanced Molecular Diagnostics (CAMD), a CLIA-certified laboratory in the Department of Pathology at Brigham and Women’s Hospital. Lesions were manually macrodissected from unstained 4 μm formalin-fixed paraffin-embedded tissue sections such that all consisted of at least 20% neoplastic nuclei. DNA was isolated by standard extraction methods with a commercially available kit (Qiagen, Valencia, CA). Genomic profiling was performed on samples with at least 50 ng/μL of DNA using an internally developed, targeted NGS panel (OncoPanel platform) as previously reported [31, 32]. The version used in this study surveyed exonic DNA sequences of 447 cancer genes and 191 regions across 60 genes for rearrangement detection. Sheared DNA was hybridized to a custom RNA bait set (Agilent SureSelect hybrid-capture kit) and sequenced on an Illumina HiSeq 2500 sequencer. Samples with a mean target coverage of <50× were excluded from the study.
Single nucleotide variants (SNVs) were called using MuTect v. 1.1.4 [33] with annotation by Oncotator [34] and indels using Indelocator (www.broadinstitute.org/cancer/cga/indelocator). Since testing was not performed on paired germline DNA, variants present at >0.1% in Exome Variant Server, NHLBI GO Exome Sequencing Project, Seattle, WA, USA (URL: http://evs.gs.washington.edu/EVS/) or gnomAD (https://gnomad.broadinstitute.org/about) were filtered and removed from analysis. Any filtered variants with an allele frequency between 0.1 and 10% were rescued and manually reviewed if found at least twice in the Catalogue of Somatic Mutations in Cancer (COSMIC; cancer.sanger.ac.uk). COSMIC was also queried to suggest functional implications of identified variants. The analysis was focused on alterations of potential biologic significance. Novel variants or those of uncertain biologic significance were reviewed and included in the analysis if evidence existed for a role of the altered gene in breast cancer, or more specifically, lobular neoplasia. Mutational burden was calculated by determining the number of nonsynonymous somatic mutations that occur per megabase of exonic sequence data across all genes on the panel.
Copy number variants (CNVs) and detection of structural variants were identified using an internally developed bioinformatics pipeline and algorithms, RobustCNV and BreaKmer [35], respectively. RobustCNV was used to calculate the fractional coverage of specified genomic intervals compared with median fractional coverage obtained in a panel of 152 FFPE nonneoplastic samples. Copy number (CN) was calculated using the formula, CN = (2 × (AGCR − 1)/P) + 2), where AGCR is the average gene copy ratio and P is the lesion purity. CN is a function of the subjective visual assessment of lesion purity, and as such, represents an estimate. An estimated CN ≥ 6 was employed as the threshold for an amplified result.
Results
Clinicopathologic features of LCIS variants
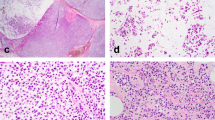
The 19 LCIS variants ranged from 0.25 to 1.0 cm in greatest linear extent (median = 0.5 cm). Of the 17 P-LCIS cases, all but one exhibited a florid growth pattern characterized by massively distended ducts and unfolded lobular acini, often densely packed and accompanied by comedo necrosis (15 cases; 88.2%). A subset displayed apocrine cytology (eight cases; 47.1%), signet ring cell features (eight cases; 47.1%), and/or a prominent stromal lymphocytic infiltrate (four cases; 23.5%) (Fig. 1). Receptor profiles included HR+/HER2− (nine cases; 52.9%), HR+/HER2+ (three cases; 17.6%), HR−/HER2+ (two cases; 11.8%), and HR−/HER2− (three cases; 17.6%). The two F-LCIS cases contained a florid growth pattern and comedo or single cell necrosis. Receptor profiles of these two cases were HR+/HER2− and HR+/HER2+. Immunohistochemical studies confirmed markedly reduced or absent membranous expression of E-cadherin and β-catenin, and aberrant cytoplasmic expression of p120 catenin in all cases included in this study, supporting a lobular phenotype.

The profiled LCIS variants represented relatively discrete lesions on biopsy that displayed a florid growth pattern with massive acinar distention (a), many with central necrosis (b). The LCIS variants met the diagnostic criteria for either florid (c) or pleomorphic (d) LCIS and some displayed apocrine (d) or signet ring cell (e) features. A single case of P-LCIS lacked marked acinar distention, and in retrospect, exhibited borderline nuclear pleomorphism (f). This case was the only one in which an ERBB2 or ERBB3 alteration was not identified.
Although all profiled biopsy specimens contained LCIS variants only, seven cases were upgraded to invasive cancer or DCIS following excision, including five cases of P-LCIS upgraded to invasive carcinoma and one case each of P-LCIS and F-LCIS upgraded to DCIS. Invasive carcinomas included: (1) Invasive lobular carcinoma (ILC), grade 2, ER+/HER2−, 0.11 cm; (2) ILC, grade 2, ER+/HER2−, 0.4 cm; (3) ILC with apocrine features, grade 2, ER+(low)/HER2−, two foci, 0.8 and 0.4 cm; (4) Invasive carcinoma with ductal and lobular features (IDLC), grade 2, ER+/HER2+, 0.15 cm; and (5) Invasive ductal carcinoma (IDC), grade 2, ER+/HER2−, 0.6 cm and microinvasive lobular carcinoma, three foci.
Genomic profile of LCIS variants
Samples were sequenced to median depth of 253× (range, 163× to 529×) with >97% of all exons >30 reads. Sequencing revealed 203 somatic mutations, including nonsynonymous SNVs and indels, affecting 121 (27.1%) of the 447 genes analyzed. The mean mutational burden was 6.884 mutations per MB (range, 2.281–10.646), which ranged from 6th to 86th percentile when compared with all breast carcinomas sequenced by this version of OncoPanel. All cases harbored three or more somatic mutations. Genes recurrently affected by mutations included CDH1, ERBB2, PIK3CA, ERBB3, RUNX1, and CBFB amongst others, with mutation frequencies of 94.7% (21 mutations in 18 cases), 68.4% (15 mutations in 13 cases), 31.6% (six mutations in six cases), 21.1% (six mutations in four cases), 21.1% (five mutations in four cases), and 15.8% (three mutations in three cases), respectively (Fig. 2 and Supplementary Data 1a).

Somatic mutations of potential biologic significance in pleomorphic and florid LCIS.
Several arm-level CN alterations were observed, including gain of 1q (16/19; 84.2%) and 16p (3/19; 15.8%), as well as loss of 16q (18/19; 94.7%), 17p (10/19; 52.6%), 8p (3/19; 15.8%), 12p (3/19; 15.8%), and 7q (2/19; 10.5%). A loss at 16q12.1-q24.3 was also present in one case (5.3%). In addition, recurrent amplifications (≥6 copies) involved ERBB2 (6/19; 31.6%), CCND1 (5/19; 26.3%), CDK12 (3/19; 15.8%), PPM1D (2/19; 10.5%), and RARA (2/19; 10.5%). GATA3, FOXA1, and AR genes were each amplified in a single case (1/19; 5.3%) (Supplementary Data 1b). Deletions of interest included a 16 bp deletion in exon 10 of CDH1 and a 780 bp deletion in RUNX1. A BAP1 (3p21.1)−5q11.1 rearrangement was the only structural rearrangement identified.
Genotype–phenotype correlation
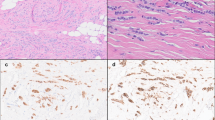
LCIS variants demonstrated biallelic inactivation of CDH1 due to concurrent LOH at 16q and CDH1 mutation in all but two cases (17/19; 89.5%). The CDH1 mutations, which consisted of 13 frameshift (13/21; 61.9%), five nonsense (5/21; 23.8%), and two missense (2/21; 9.5%) mutations, as well as one 16-base pair deletion (1/21; 4.8%), were distributed throughout the coding sequence in multiple domains of the gene (Fig. 3 and Supplementary Data 1a). All mutations were consistent with loss-of-function, except for one missense variant of uncertain significance. These findings provide further evidence that these lesions have a lobular phenotype.

Biallelic inactivation of the CDH1 gene occurred via concurrent 16q loss and CDH1 mutation in the majority (17/19) cases (a). CDH1 mutations were scattered throughout the coding sequence and included frameshift, nonsense, and missense mutations as well as a 16-base pair deletion (b).
The LCIS variants were otherwise characterized by highly recurrent alterations in ERBB2 (HER2) and ERBB3 (HER3), members of the epidermal growth factor receptor family of receptor tyrosine kinases. ERBB2 and/or ERBB3 alterations were found in 18 of 19 (94.7%) cases, including both F-LCIS cases. ERBB2 mutations consisted of missense mutations clustered in the tyrosine kinase and furin-like extracellular domains as well as insertions in the tyrosine kinase domain (Fig. 4 and Supplementary Data 1a). ERBB2 mutations were predominantly activating; however, p.L755S (c.2264T>C), the most common mutation (seven cases; 36.8%), is known to confer lapatinib resistance, but its effect on kinase activity is uncertain (COSMIC v90 and Bose et al. [36]). ERBB3 mutations were missense mutations found in the tyrosine kinase, furin-like cysteine rich, and receptor L domains. Four of five mutations were previously reported in multiple breast cancer samples and predicted to be pathogenic, whereas a single variant in the kinase domain was novel (Supplementary Data 1a). There were seven cases with concurrent ERBB2 and/or ERBB3 alterations, including ERBB2 mutation and amplification (two cases), two different ERBB2 mutations (two cases), and ERBB2 and ERBB3 mutations (three cases) (Fig. 4a). ERBB2 mutant allele frequencies greater than estimated tumor cellularity in the two cases with concurrent amplification are consistent with alteration of the same allele. All cases of LCIS with florid growth pattern (18 cases) or apocrine features (eight cases) had ERBB2 or ERBB3 alterations. Of six cases with ERBB2 amplification, five cases (7 to 34 estimated copies) were interpreted as positive (3+) by immunohistochemistry, while a single case (eight estimated copies) was negative (0–1+).

ERRB2 and/or ERBB3 alterations were observed in all but one case (a), with two or more concurrent alterations in a subset. ERBB2 mutations were predominantly missense mutations found within the protein tyrosine kinase and furin-like extracellular domains and predicted to be activating (b). Amp amplification, Mut mutation, ECD extracellular domain, TM transmembrane region, PTK protein tyrosine kinase.
Apart from this, no other significant correlations were noted between genomic alterations and morphologic features or upgrade on excision (Fig. 5), although it is worth noting that two cases represented outliers in our study. First, the only case that lacked ERBB2 and ERBB3 alterations was a case of HR+/HER2− P-LCIS with distinct morphologic features. This case, which was associated with invasive carcinoma on excision, did not exhibit the florid growth pattern nor the same degree of nuclear pleomorphism as seen in the other P-LCIS cases, but rather featured a prominent stromal lymphocytic infiltrate (Fig. 1f). It harbored 1q gain and 16q loss without concurrent CDH1 mutation or other explanation for biallelic inactivation of CDH1. CDH1 promoter methylation was not investigated. The second outlier, a case of HR−/HER2+ pure P-LCIS with necrosis, was morphologically similar to the other cases yet contained a single TP53 mutation (nonsense, pE56*). Moreover, although this case was E-cadherin-negative and also harbored a CDH1 mutation, neither 1q gain nor 16q loss were present and the mechanism of biallelic CDH1 inactivation remains uncertain. CDH1 promoter methylation was not investigated. The findings raise the possibility of a distinct neoplasia pathway.

All cases with a florid growth pattern or apocrine features harbored the latter. Outlier cases included (1) HER2+ P-LCIS with a TP53 mutation and no 16q loss or 1q gain, and (2) ER+ P-LCIS that lacked a florid growth pattern, as well as CDH1 and ERBB2 or ERBB3 mutations. No other significant correlations were noted between genomic alterations and morphologic features or upgrade to malignancy. LCIS lobular carcinoma in situ, DCIS ductal carcinoma in situ, HR hormone receptor, HER2 human epidermal growth factor receptor 2.
Discussion
Herein, we report results of genomic profiling of a large series of LCIS variants diagnosed on core biopsies at a single institution using our internally-developed targeted NGS assay (OncoPanel). Our study revealed that these LCIS variants, which were all confirmed as E-cadherin and β-catenin negative and p120 aberrant, harbor genomic alterations consistent with a lobular phenotype, most importantly biallelic inactivation of the CDH1 gene by concurrent 16q loss and CDH1 mutation, as well as recurrent mutations in PIK3CA, RUNX1, and CBFB. Moreover, the LCIS variants, particularly those with a florid growth pattern, are characterized by highly recurrent ERBB2 and ERBB3 alterations not generally seen in C-LCIS, suggesting that signaling via the epidermal growth factor receptor family of receptor tyrosine kinases is an important oncogenic driver in the development of these lesions. CCND1 amplification, which is not only recurrent in our study, but also has been previously reported as enriched in LCIS variants compared with C-LCIS, may also play a role in a subset of cases.
Much of what was previously known about the molecular profile of LCIS and its morphologic variants came from single gene mutation, loss of heterozygosity (LOH) and DNA CN analyses by low-throughput and/or low-resolution approaches published several years ago. From these studies, it was established that C-LCIS shares a similar pattern of chromosomal alterations and low-level genetic complexity as other lesions in the ER-positive low-grade breast neoplasia family (e.g., flat epithelial atypia, atypical ductal hyperplasia, low-grade DCIS, well-differentiated IDCs, and classic ILCs), including recurrent gains of 1q and losses of 16q and 17p [37,38,39,40,41,42]. Array-based CGH studies of LCIS variants [15, 43, 44], which had previously represented the best efforts to molecularly characterize these lesions, subsequently demonstrated that P-LCIS and F-LCIS consistently display 1q gain and 16q loss as well. However, apocrine P-LCIS and F-LCIS contain more genomic alterations than nonapocrine P-LCIS and C-LCIS, with the former enriched for amplification of 17q11.2-17q12 (the region harboring the HER2 gene), amplification of 11q13.3 (the region harboring the cyclin D1 gene), gain of 16p, and losses of 3q, 11q, and 13q. The findings suggested that at least some LCIS variants represent more genetically advanced forms of LCIS, although it should be noted that the degree of genetic instability in these lesions is still less than that seen in high-grade DCIS.
More recently, massively parallel sequencing has enabled more comprehensive molecular profiling and provided greater insight into the biology of invasive and in situ lobular neoplasia of the breast. For example, our understanding of the lobular phenotype has been enhanced by profiling of ILC by the Cancer Genome Atlas (TCGA) Research Network. In a study by Ciriello et al. [45] 817 breast tumor samples, including 127 ILCs, 490 IDCs, and 88 IDLCs, were subjected to whole-exome DNA sequencing amongst other molecular assays. ILC cases, predominantly ER+ and luminal A, were significantly enriched for mutations affecting CDH1 (63% in ILC vs. 2% in IDC), PIK3CA (48% vs. 33%), RUNX1 (10% vs. 3%), TBX3 (9% vs. 2%), and FOXA1 (7% vs. 2%). Alterations characteristic of ER-/basal-like tumors were less frequent in ILC than IDC, including TP53 mutations (8% in ILC vs. 44% in IDC) and focal amplification of MYC (6% vs. 27%) and CCNE1 (0% vs. 7%). CDH1 mutations were uniformly distributed along the coding sequence, mostly truncating (83%), and almost invariably cooccurring with heterozygous loss of 16q. Although previous studies had suggested epigenetic silencing as a mechanism of CDH1 downregulation, significant DNA hypermethylation of the CDH1 promoter region was not detected. The ERBB2 mutation rate in this and a prior TCGA study [46] was ~2%; however, the relative representation of ILC with “classic” versus “pleomorphic” morphology in these studies was not specified.
NGS-based genomic profiling studies of paired C-LCIS and ILC revealed a similar repertoire of somatic mutations as described in the TCGA study. Sakr et al. [47] subjected fresh frozen and microdissected samples with LCIS and ILC to massively parallel sequencing targeting exons of 273 genes. LCIS and ILC harbored a similar constellation of somatic mutations, with the most frequently mutated genes being CDH1 (56% and 66%, respectively), PIK3CA (41% and 52%, respectively), and CBFB (12% and 19%, respectively). TP53 mutations were rare (two cases; 6%) and ERBB2 mutations were not reported with other recurrently mutated genes. In LCIS, CDH1 mutations, which were distributed across multiple domains of the gene and considered nonpassenger events, cooccurred with LOH at 16q in 18 of 19 samples. In another study by the same group [48], whole-exome sequencing once again revealed that the most frequently recurring mutations involved CDH1 and PIK3CA, followed by CBFB and GATA3, amongst others. Rare ERBB2 (three V777L missense mutations) and no TP53 mutations were reported in LCIS. Our findings of recurrent mutations in CDH1, PIK3CA, CBFB, and RUNX1 in LCIS variants is consistent with the lobular profile suggested by these studies of C-LCIS and ILC, whereas highly recurrent ERBB2 and ERBB3 mutations appear to represent a finding more specific to pleomorphic and/or florid lesions.
Similar conclusions can be drawn from studies that have sequenced pleomorphic invasive lobular carcinoma (P-ILC), a variant with more aggressive features than classic ILC (C-ILC). In a recent study of 37 cases of P-ILC using a custom 34-gene panel by Rosa-Rosa et al. [49], CDH1 (89%), PIK3CA (33%), and ERBB2 (26%) were the most commonly mutated genes. Alterations related to progression from in situ to invasive carcinoma and/or lymph node metastasis included TP53 mutation and amplification of PIK3CA and CCND1; however, the histologic subtypes of the seven profiled LCIS lesions were not specified. ERBB2 mutations were noted in both components of two cases of synchronous LCIS and P-ILC. Similarly, another study of 17 cases of P-ILC subjected to targeted sequencing showed that P-ILC contained many of the alterations characteristic of ILC, but was enriched for amplification or mutation of ERBB2 (11.8% and 17.6%, respectively) and ERBB3 (5.9% and 23.5%, respectively) [50]. According to prior work by Lien et al. [51], the frequency of ERBB2 mutations and overexpression/amplification in pleomorphic ILC (11 of 21 cases; 52%) and LCIS (2 of 3 cases; 67%) may be even higher.
It should be acknowledged that our study is not the only one to apply an NGS-based approach to investigate the genomic profile of pleomorphic and florid variants of LCIS. In a recent abstract, Shamir et al. [52] reported results from targeted capture-based NGS profiling of nine cases of synchronous C-LCIS, LCIS variants (five P-LCIS and four F-LCIS), and ILC, as well as five cases of pure LCIS variants (three P-LCIS and two F-LCIS). Almost all cases displayed shared pathogenic mutations and CNVs between LCIS variants and ILC and between LCIS variants/ILC and C-LCIS. Recurrent genomic alterations in LCIS variants included mutations in CDH1, PIK3CA, ERBB2, ERBB3, and TP53 and amplification of CCND1, with ERBB2 and/or ERBB3 mutations present in half of the cases of P-LCIS and F-LCIS as well as two cases of C-LCIS. This mutational profile is strikingly similar to that seen in our own series, with enrichment of ERBB2 and ERBB3 mutations representing the most compelling finding. Taken together, several studies of P-ILC and P-LCIS have provided a growing body of evidence implicating ERBB2 alterations and presumably epidermal growth factor receptor family-related tyrosine kinase signaling in the pathogenesis of lobular neoplasia with pleomorphic morphologic features.
Our study and the other studies reviewed herein suggest a plausible theory regarding the pathogenesis of pleomorphic and florid LCIS, in which many of these lesions regardless of their HR status likely evolve from classic lobular neoplasia, with subsequent acquisition of additional pathogenic alterations, including ERBB2 or ERBB3 mutation or amplification, as well as various chromosomal arm-level CN alterations more frequently reported in high-grade carcinomas. In the commonly accepted model of breast cancer evolution, lesions of the low-grade neoplasia pathway are HR+/HER2− and display a simple karyotype and concurrent deletion of 16q and gains of 1q and 16p, whereas those of the high-grade neoplasia pathway, which may be HR− or HER2+, tend to have complex karyotypes with losses of 1p, 8p, and 17p and gains of 1q and 8q, amongst others [39]. Loss of 16q, the hallmark genetic alteration of low-grade breast cancers, is observed in <30% of high-grade lesions, which has led to the hypothesis that progression from low- to high-grade breast cancer is an uncommon biological phenomenon [49, 53]. In accordance, high-grade DCIS, which infrequently contains arm-level loss of 16q, is thought in a majority of cases to arise de novo or from a precursor other than atypical ductal hyperplasia and/or low-grade DCIS [39]. P-LCIS appears to represent an exception to this simple two-pathway model. Despite increased genetic complexity and some shared genomic alterations with the high-grade pathway, P-LCIS almost always harbors concurrent 16q loss and 1q gain and other mutations seen in classic lobular neoplasia. A single case in our series that lacked concurrent 16q loss and 1q gain and displayed a TP53 mutation suggests that an alternative neoplasia pathway may exist in a small subset of cases, with P-LCIS either arising de novo, or perhaps from high-grade DCIS, with biallelic loss of E-cadherin due to other mechanisms at a later stage in evolution.
Again, the most significant finding in our study and one that sets it apart from other studies of pleomorphic lobular neoplasia, is the high frequency of ERBB2 and ERBB3 alterations in LCIS variants (18 of 19 cases; 94.7%), including ERBB2 or ERBB3 mutations in 12 out of 13 (92.3%) cases classified as negative or equivocal by immunohistochemistry. Our findings may be related to our approach to case selection, which may be considered both a strength and limitation of this study. Although we began by reviewing consecutive cases of LCIS with variant morphology diagnosed at our institution over a greater than 10-year period, we selected cases for genomic profiling with relatively discrete, florid lesions containing closely packed, and massively distended acini that represented the targeted radiologic lesion on core needle biopsy. We believe that the extent, cellularity, and purity of these lesions facilitated technically successful sequencing and biologically representative results. However, our selection bias toward florid lesions cannot be ignored, and it is possible that ERBB2 and ERBB3 alterations are particularly enriched in florid lesions. It is notable that ERBB2 and ERBB3 alterations were not found in the only case that lacked a florid growth pattern in our series. Another contributing factor may be that apocrine features were appreciated in many of the selected cases and this type of morphology has been associated with HER2 signaling [54]. Although ERBB2 alterations were found in both F-LCIS cases, the small number of F-LCIS cases in our series limits further conclusions regarding the molecular phenotype specific to this subtype.
In summary, genomic profiling of pleomorphic and florid variants of LCIS confirms that these lesions represent a form of lobular neoplasia, albeit more genetically advanced than lesions with classic morphology, and suggests that phenotypic variation may be driven by genomic alterations that upregulate HER2 signaling. Our results, in conjunction with those of other studies, support the distinction of these LCIS variants from C-LCIS in the current WHO classification. Furthermore, they suggest that P-LCIS is also biologically distinct from predominant forms of high-grade DCIS, highlighting the need to further study the natural history of these lesions as opposed to extrapolating DCIS management approaches to P-LCIS. It is hoped that more uniform adoption of new WHO diagnostic criteria will facilitate collection the data needed to better inform clinical management.
References
Foote FW, Stewart FW. Lobular carcinoma in situ: a rare form of mammary cancer. Am J Pathol. 1941;17:491–6.3.
Haagensen CD, Lane N, Lattes R, Bodian C. Lobular neoplasia (so-called lobular carcinoma in situ) of the breast. Cancer. 1978;42:737–69.
Dabbs DJ, Schnitt SJ, Geyer FC, Weigelt B, Baehner FL, Decker T, et al. Lobular neoplasia of the breast revisited with emphasis on the role of E-cadherin immunohistochemistry. Am J Surg Pathol. 2013;37:e1–11.
Canas-Marques R, Schnitt SJ. E-cadherin immunohistochemistry in breast pathology: uses and pitfalls. Histopathology. 2016;68:57–69.
National Comprehensive Cancer Network (US) Clinical practice guidelines in oncology, breast cancer (version 3.2019). Fort Washington, PA: National Comprehensive Cancer Network (US); 2019. https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed on 11/02/2019.
National Comprehensive Cancer Network (US) Clinical practice guidelines in oncology, breast cancer screening and diagnosis (version 1.2019). Fort Washington, PA: National Comprehensive Cancer Network (US); 2019. https://www.nccn.org/professionals/physician_gls/pdf/breast_screening.pdf. Accessed on 11/02/2019.
Brogi E, Murray MP, Corben AD. Lobular carcinoma, not only a classic. Breast J. 2010;16 Suppl 1:S10–4.
Murray M, Brogi E. Lobular carcinoma in situ, classical type and unusual variants. Surg Pathol Clin. 2009;2:273–99.
Lakhani SR, Schnitt SJ, O’Malley F, Van de Vijver MJ, Simpson PT, Palacios J. Lobular neoplasia. In: Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJ, editors. WHO classification of tumours of the breast. 4th ed. Lyon, France: International Agency for Research on Cancer; 2012.
Fadare O, Dadmanesh F, Alvarado-Cabrero I, Snyder R, Stephen Mitchell J, Tot T, et al. Lobular intraepithelial neoplasia [lobular carcinoma in situ] with comedo-type necrosis: a clinicopathologic study of 18 cases. Am J Surg Pathol. 2006;30:1445–53.
Chen YY, Decker T, King TA, Palacios J, Shin SJ, Simpson PT. Lobular carcinoma in situ. In: WHO classification of tumours editorial board. WHO classification of tumours of the breast. 5th ed. Lyon, France: International Agency for Research on Cancer; 2019.
Shamir ER, Chen YY, Chu T, Pekmezci M, Rabban JT, Krings G. Pleomorphic and florid lobular carcinoma in situ variants of the breast: a clinicopathologic study of 85 cases with and without invasive carcinoma from a single academic center. Am J Surg Pathol. 2019;43:399–408.
Wen HY, Brogi E. Lobular carcinoma in situ. Surg Pathol Clin. 2018;11:123–45.
Alvarado-Cabrero I, Picon Coronel G, Valencia Cedillo R, Canedo N, Tavassoli FA. Florid lobular intraepithelial neoplasia with signet ring cells, central necrosis and calcifications: a clinicopathological and immunohistochemical analysis of ten cases associated with invasive lobular carcinoma. Arch Med Res. 2010;41:436–41.
Chen YY, Hwang ES, Roy R, DeVries S, Anderson J, Wa C, et al. Genetic and phenotypic characteristics of pleomorphic lobular carcinoma in situ of the breast. Am J Surg Pathol. 2009;33:1683–94.
Nakhlis F, Harrison BT, Giess CS, Lester SC, Hughes KS, Coopey SB, et al. Evaluating the rate of upgrade to invasive breast cancer and/or ductal carcinoma in situ following a core biopsy diagnosis of non-classic lobular carcinoma in situ. Ann Surg Oncol. 2019;26:55–61.
Chivukula M, Haynik DM, Brufsky A, Carter G, Dabbs DJ. Pleomorphic lobular carcinoma in situ (PLCIS) on breast core needle biopsies: clinical significance and immunoprofile. Am J Surg Pathol. 2008;32:1721–6.
Carder PJ, Shaaban A, Alizadeh Y, Kumarasuwamy V, Liston JC, Sharma N. Screen-detected pleomorphic lobular carcinoma in situ (PLCIS): risk of concurrent invasive malignancy following a core biopsy diagnosis. Histopathology. 2010;57:472–8.
Guo T, Wang Y, Shapiro N, Fineberg S. Pleomorphic lobular carcinoma in situ diagnosed by breast core biopsy: clinicopathologic features and correlation with subsequent excision. Clin Breast Cancer. 2018;18:e449–54.
Murray MP, Luedtke C, Liberman L, Nehhozina T, Akram M, Brogi E. Classic lobular carcinoma in situ and atypical lobular hyperplasia at percutaneous breast core biopsy: outcomes of prospective excision. Cancer. 2013;119:1073–9.
Nakhlis F, Gilmore L, Gelman R, Bedrosian I, Ludwig K, Hwang ES, et al. Incidence of adjacent synchronous invasive carcinoma and/or ductal carcinoma in-situ in patients with lobular neoplasia on core biopsy: results from a prospective multi-institutional registry (TBCRC 020). Ann Surg Oncol. 2016;23:722–8.
Susnik B, Day D, Abeln E, Bowman T, Krueger J, Swenson KK, et al. Surgical outcomes of lobular neoplasia diagnosed in core biopsy: prospective study of 316 cases. Clin Breast Cancer. 2016;16:507–13.
Chaudhary S, Lawrence L, McGinty G, Kostroff K, Bhuiya T. Classic lobular neoplasia on core biopsy: a clinical and radio-pathologic correlation study with follow-up excision biopsy. Mod Pathol. 2013;26:762–71.
Downs-Kelly E, Bell D, Perkins GH, Sneige N, Middleton LP. Clinical implications of margin involvement by pleomorphic lobular carcinoma in situ. Arch Pathol Lab Med. 2011;135:737–43.
Sneige N, Wang J, Baker BA, Krishnamurthy S, Middleton LP. Clinical, histopathologic, and biologic features of pleomorphic lobular (ductal-lobular) carcinoma in situ of the breast: a report of 24 cases. Mod Pathol. 2002;15:1044–50.
Khoury T, Karabakhtsian RG, Mattson D, Yan L, Syriac S, Habib F, et al. Pleomorphic lobular carcinoma in situ of the breast: clinicopathological review of 47 cases. Histopathology. 2014;64:981–93.
De Brot M, Koslow Mautner S, Muhsen S, Andrade VP, Mamtani A, Murray M, et al. Pleomorphic lobular carcinoma in situ of the breast: a single institution experience with clinical follow-up and centralized pathology review. Breast Cancer Res Treat. 2017;165:411–20.
Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013.
Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36:2105–22.
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Pathol Lab Med. 2010;134:e48–72.
Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of oncopanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–8.
Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9.
Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, et al. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–9.
Abo RP, Ducar M, Garcia EP, Thorner AR, Rojas-Rudilla V, Lin L, et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 2015;43:e19.
Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–37.
Lakhani SR, Collins N, Sloane JP, Stratton MR. Loss of heterozygosity in lobular carcinoma in situ of the breast. Clin Mol Pathol. 1995;48:M74–8.
Lu YJ, Osin P, Lakhani SR, Di Palma S, Gusterson BA, Shipley JM. Comparative genomic hybridization analysis of lobular carcinoma in situ and atypical lobular hyperplasia and potential roles for gains and losses of genetic material in breast neoplasia. Cancer Res. 1998;58:4721–7.
Lopez-Garcia MA, Geyer FC, Lacroix-Triki M, Marchio C, Reis-Filho JS. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology. 2010;57:171–92.
Bombonati A, Sgroi DC. The molecular pathology of breast cancer progression. J Pathol. 2011;223:307–17.
Morandi L, Marucci G, Foschini MP, Cattani MG, Pession A, Riva C, et al. Genetic similarities and differences between lobular in situ neoplasia (LN) and invasive lobular carcinoma of the breast. Virchows Arch. 2006;449:14–23.
Mastracci TL, Shadeo A, Colby SM, Tuck AB, O’Malley FP, Bull SB, et al. Genomic alterations in lobular neoplasia: a microarray comparative genomic hybridization signature for early neoplastic proliferationin the breast. Genes Chromosomes Cancer. 2006;45:1007–17.
Boldt V, Stacher E, Halbwedl I, Popper H, Hultschig C, Moinfar F, et al. Positioning of necrotic lobular intraepithelial neoplasias (LIN, grade 3) within the sequence of breast carcinoma progression. Genes Chromosomes Cancer. 2010;49:463–70.
Shin SJ, Lal A, De Vries S, Suzuki J, Roy R, Hwang ES, et al. Florid lobular carcinoma in situ: molecular profiling and comparison to classic lobular carcinoma in situ and pleomorphic lobular carcinoma in situ. Hum Pathol. 2013;44:1998–2009.
Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–19.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Sakr RA, Schizas M, Carniello JV, Ng CK, Piscuoglio S, Giri D, et al. Targeted capture massively parallel sequencing analysis of LCIS and invasive lobular cancer: repertoire of somatic genetic alterations and clonal relationships. Mol Oncol. 2016;10:360–70.
Begg CB, Ostrovnaya I, Carniello JV, Sakr RA, Giri D, Towers R, et al. Clonal relationships between lobular carcinoma in situ and other breast malignancies. Breast Cancer Res. 2016;18:66.
Rosa-Rosa JM, Caniego-Casas T, Leskela S, Cristobal E, Gonzalez-Martinez S, Moreno-Moreno E, et al. High Frequency of ERBB2 activating mutations in invasive lobular breast carcinoma with pleomorphic features. Cancers. 2019;11:1–16.
Zhu S, Ward BM, Yu J, Matthew-Onabanjo AN, Janusis J, Hsieh CC, et al. IRS2 mutations linked to invasion in pleomorphic invasive lobular carcinoma. JCI Insight. 2018;3:1–11.
Lien HC, Chen YL, Juang YL, Jeng YM. Frequent alterations of HER2 through mutation, amplification, or overexpression in pleomorphic lobular carcinoma of the breast. Breast Cancer Res Treat. 2015;150:447–55.
Shamir E, Chu T, Chen YY, Krings G. Genomic profiling of lobular carcinoma in situ (LCIS) variants with comparison to classic lcis and invasive lobular carcinoma (abs. #. 295). Mod Pathol. 2018;31 Suppl 2:104.
Simpson PT, Reis-Filho JS, Gale T, Lakhani SR. Molecular evolution of breast cancer. J Pathol. 2005;205:248–54.
Bhargava R, Beriwal S, Striebel JM, Dabbs DJ. Breast cancer molecular class ERBB2: preponderance of tumors with apocrine differentiation and expression of basal phenotype markers CK5, CK5/6, and EGFR. Appl Immunohistochem Mol Morphol. 2010;18:113–8.
Acknowledgements
We would like to acknowledge the CAMD of Brigham and Women’s Hospital for their technical contributions to this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
DAD consults for Novartis and is on the Academic Advisory Board of Oncology Analytics, Inc. The other authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Harrison, B.T., Nakhlis, F., Dillon, D.A. et al. Genomic profiling of pleomorphic and florid lobular carcinoma in situ reveals highly recurrent ERBB2 and ERRB3 alterations. Mod Pathol 33, 1287–1297 (2020). https://doi.org/10.1038/s41379-020-0459-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0459-6
This article is cited by
-
Invasive lobular carcinoma with extracellular mucin (ILCEM): clinicopathologic and molecular characterization of a rare entity
Modern Pathology (2022)
-
Current Perspectives on Lobular Neoplasia of the Breast
Current Radiology Reports (2022)
-
The morphologic spectrum of lobular carcinoma in situ (LCIS) observations on clinical significance, management implications and diagnostic pitfalls of classic, florid and pleomorphic LCIS
Virchows Archiv (2022)
-
Invasive lobular carcinoma of the breast: the increasing importance of this special subtype
Breast Cancer Research (2021)
-
Morphologic subtypes of lobular carcinoma in situ diagnosed on core needle biopsy: clinicopathologic features and findings at follow-up excision
Modern Pathology (2021)
