
Abstract
Sporadic reports of t(3;12)(q26.2;p13) indicate that this abnormality is associated with myeloid neoplasms, myelodysplasia, and a poor prognosis. To better characterize neoplasms with this abnormality, we assessed 20 patients utilizing clinicopathological data, cytogenetic, and targeted next-generation sequencing analysis. We also performed literature review of 58 prior reported cases. Patients included ten men and ten women with median age 55.8 years (range, 27.8–78.8). Diagnoses included 11 acute myeloid leukemia (AML, 5 de novo and 6 secondary), 5 myelodysplastic syndromes (MDS, 3 de novo excess blasts-2 and 2 therapy-related), 2 chronic myeloid leukemia BCR-ABL1-positive blast phase (1 de novo and 1 secondary), 1 primary myelofibrosis (secondary), and 1 mixed-phenotype acute leukemia T/myeloid (MPAL, secondary). Morphologic dysplasia was identified in all AML cases (5/5), MDS cases (4/4), therapy-related cases (3/3), half of myeloproliferative neoplasm cases (1/2), and one MPAL case assessed. The t(3;12) was detected de novo and in subsequent workups in 9 and 11 patients, respectively. Seven patients had t(3;12) only and eight patients had additional chromosome 7 abnormalities. Fluorescence in-situ hybridization detected MECOM (n = 11) and ETV6 (n = 7) rearrangements in all cases assessed. FLT3 internal tandem duplication was identified in five (25%) patients. We identified 13 genetic abnormalities in the de novo group (n = 9), and 25 in the secondary disease group (n = 11). All patients received chemotherapy, with seven allogeneic and two autologous stem cell transplantations. At last follow-up, 14 (70%) patients died with median survival of 6.3 months (range, 0.1–17.3) after detection of t(3;12). In summary, t(3;12)(q26.2;p13) is a rare cytogenetic abnormality in myeloid neoplasms. Myelodysplasia, chromosome 7 abnormalities, and high blast counts are common, and the prognosis is poor. Given the close relationship between the presence of this cytogenetic abnormality and the MDS-related changes, we recommend adding t(3;12)(q26.2;p13) to the list of AML with myelodysplasia-related changes defining abnormalities of the World Health Organization 2017 classification of myeloid neoplasms.
Similar content being viewed by others
Introduction
The MECOM locus consists of the MYELODYSPLASTIC SYNDROME 1 (MDS1) and ecotropic viral integration site-1 (EVI1) genes located at 3q26.2. EVI1 was first identified as a pro-viral insertion site in murine leukemia [1]. MECOM/3q26.2 rearrangement has been observed in various myeloid neoplasms, such as acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), myelodysplastic/myeloproliferative neoplasms, and chronic myeloid leukemias [2,3,4,5,6]. Patients with myeloid neoplasms associated with MECOM/3q26 abnormalities have a poor response to chemotherapy and a poor outcome.
MECOM/3q26 abnormalities result in overexpression of MECOM. Overexpression can be achieved via two mechanisms. The first mechanism involves juxtaposition of regulatory sequences near MECOM as a result of inv(3)(q21q26), t(3;3)(q21;q26), t(3;7)(q26;q21), or t(3;17)(q26;q22) [7,8,9,10,11]. The second mechanism results in fusion of the regulatory sequences near the MECOM gene as occurs in t(3;21)(q26;q22) and t(3;12)(q26;p13) where MECOM fuses with RUNX1 or ETV6 (formerly called TEL), respectively [7, 12]. Overexpression of MECOM as a result of MECOM/ETV6 fusion is driven by the ETV6 promoter resulting in cell proliferation and malignant transformation [13]. However, the mechanism by which MECOM induces leukemogenesis is not well understood.
Although myeloid neoplasms associated with MECOM abnormalities have been reported, in most of these studies the t(3;21)(q26;q22) or inv(3)(q21q26) were the focus. Myeloid neoplasms associated with t(3;12)(q26.2;p13) have received very little attention in the literature, mostly as case reports, rare case series, or as a minor mention in larger studies [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40]. In this study, we characterize the clinicopathological features of 20 patients with myeloid neoplasms associated with t(3;12)(q26.2;p13).
Materials and methods
Case collection
We searched the files at our institution from January 1, 2000 through December 31, 2018 for hematologic neoplasms assessed by conventional cytogenetics and shown to carry t(3;12)(q26.2;p13). The following data were collected and analyzed: clinicopathological parameters, treatment regimens, response, and follow-up data. This study was approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center.
We reviewed peripheral blood smears, bone marrow (BM) aspirate smears, and touch imprints that were stained with Wright–Giemsa. We also reviewed aspirate clot specimens and decalcified BM core biopsy specimens stained with hematoxylin and eosin.
Cytogenetic analysis
Conventional cytogenetic analyses were performed on G-banded metaphase cells using standard methods. Twenty metaphases were analyzed and the results were reported according to the current International System for Human Cytogenetic Nomenclature. Fluorescence in-situ hybridization (FISH) analysis was performed on cultured BM cells or air-dried aspirate smears following standard protocols. The presence of t(3;12)(q26.2;p13) was confirmed by FISH analysis to involve either MECOM (Leica/Kreatech Inc.), ETV6 (Abbott), or both using a dual color, break-apart DNA probe. A total of 200 interphase nuclei were analyzed.
Mutation analysis
Next-generation sequencing analysis using a 28- or 81-gene panel was performed on ten cases following the instructions described previously [41]. Mutation analysis for PTPN11, GATA binding protein 2 (GATA2), KRAS, NRAS, and RUNX1 was included in both gene mutation panels. In the remaining ten cases, PCR-based DNA Sanger sequencing and reverse transcriptase PCR were performed to assess the following genes: C-KIT, NRAS, KRAS, FLT3, NPM1, CEBPA, IDH1, IDH2, DNMT3A, and JAK2. For all 20 cases, reverse transcriptase PCR was used to assess for BCR-ABL1
Results
Clinicopathological characteristics
Patients included ten men and ten women with a median age of 55.8 years (range, 27.8–78.8) at time of initial diagnosis (Table 1). The t(3;12)(q26.2;p13) was detected at time of initial diagnosis in 9 patients and was detected over the clinical course in the other 11 patients. At t(3;12) detection, morphologic myelodysplasia was observed in the majority of patients (11 of 12, 92%) assessed (Fig. 1). One patient did not initially show myelodysplasia, but demonstrated myelodysplasia during the disease course (case #1). Of the remaining eight patients that precluded assessment of myelodysplasia at time of t(3;12) detection, six had too few cells in the aspirates and/or touch imprints for assessment but showed myelodysplasia at a later time point. The other two cases were too limited in quality, precluding reliable assessment. All three therapy-related cases showed evidence of dysplasia (one t-AML and two t-MDS).

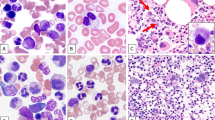
a A representative case of BM core biopsy of AML with t(3;12)(q26.2;p13) (case #11) with a 100% hypercellular marrow with a cluster of dysplastic megakaryocytes, three of which are highlighted by arrowheads. b Aspirate shows myelodysplasia in erythroid precursors (top left: irregular nuclear contours; top right: irregular nuclear contours and cytoplasmic vacuolization; bottom left: nuclear cytoplasmic asynchrony) and megakaryocytes (bottom right: hypolobated). c Aspirate shows myelodysplasia in granulocytic precursors (top left: irregular nuclear contours and hypogranularity; top right: irregular nuclear contours; bottom left: pseudo-Pelger-Huet form) and megakaryocytes (bottom right: micro-megakaryocyte). d A representative case of BM core biopsy of therapy-related MDS with t(3;12)(q26.2;p13) (case #13) with a 30% cellular marrow. e Aspirate shows dysplasia in erythroid precursors with nuclear: cytoplasmic asynchrony and mildly irregular nuclear contours (arrowhead); dysplasia in granulocytic precursors with abnormal nuclear segmentation and abnormally clumped chromatin (arrows) as well as and pseudo-Pelger-Huet form granulocyte (inset). f Dysplastic megakaryocytes with separated nuclear lobes as well as the presence of a representative ring sideroblast (inset). g A representative BM core biopsy of CML-BP (CML, BCR-ABL1-positive, BP) with t(3;12)(q26.2;p13) (case #7) with a hypercellular marrow with extensive myelofibrosis. h Aspirate with increased blasts. i Reticulin stain demonstrates increased myelofibrosis, trichrome stain showed a few scattered collagen fibers (not shown) giving an overall grade of MF-2 (scale of MF-0–MF-3). Original magnification: a ×400; d, g, i ×200; e, f, h ×400; b, c, inset ×1000.
At the time t(3;12) was detected, 11 patients had AML and 1 patient had mixed-phenotype acute leukemia T/myeloid not otherwise specified (MPAL), per World Health Organization 2017 (WHO 2017) classification of myeloid neoplasms. These patients consisted of AML with myelodysplasia-related changes (AML-MRC, n = 8), acute monoblastic leukemia (n = 1), not otherwise specified (n = 1), therapy related (n = 1), and MPAL (n = 1). The t-AML patient had plasma cell myeloma (treated with bortezomib plus dexamethasone) and developed lung carcinoma 4 years later (treated with radiation). The myeloma was treated with autologous stem cell transplant (SCT) 6 months after the radiation. Five years after SCT, he developed t-MDS with 7% blasts. Twenty months after the MDS diagnosis, his disease evolved to t-AML with t(3;12) and 40% BM blasts. Of the 11 AML cases, 5 arose de novo and 6 were secondary-not including the secondary MPAL case. The MPAL patient initially had AML and evolved, as defined as progression in disease without prior complete remission, with phenotypic shift to MPAL after 25.4 months. In the AML group, the interval from initial disease to AML ranged from 0 to 10.5 years. For the eight AML patients with available data, the median BM blasts were 48% (range, 17–91%). Complete blood count (CBC) in these patients showed blasts 26% (range, 0–88%), WBC 14.1 K/µl (range, 0.3–129.5), Hgb 8.3 g/dl (range, 6.8–10.3), and platelets 61 K/µl (range, 25–186). At t(3;12) detection, all of the five AML patients assessed (100%) demonstrated dysplasia as did the one MPAL patient. The dysplastic features identified in this group are as follows: granulocytic precursors (irregular nuclear contours including pseudo-Pelger-Huet forms and cytoplasmic hypogranularity), erythroid precursors (irregular nuclear contours including bi-nucleation and nuclear budding, cytoplasmic vacuoles, and nuclear cytoplasmic asynchrony), megakaryocytes (small, nuclear hypolobation, micro-megakaryocytes). At time of t(3;12) detection, three AML and one MPAL patient had myelodysplasia in all three lineages, while one t-AML patient had myelodysplasia in two lineages, and one AML-MRC had myelodysplasia in one lineage. Six AML cases were not assessed for dysplasia due to too few non-blast cells. The findings are summarized in Table 1.
At the time t(3;12) was detected, the five MDS cases included three de novo and two therapy-related disease. In the latter group, one patient was treated for diffuse large B-cell lymphoma, 52 months later a BM biopsy performed due to cytopenia showed MDS with t(3;12), 1% blasts in a 50% cellular aspirate clot with myelodysplasia in the granulocytic and the erythroid precursors; megakaryocytes were markedly decreased in numbers. The other t-MDS patient was treated for breast cancer 12 years prior to development of t-MDS. The five MDS cases showed median BM blasts of 10% (range, 1–18%). The CBC showed blasts 1% (range, 0–2%), WBC 1.9 K/µl (range, 1.8–4.0), Hgb 9.8 g/dl (range, 7.7–11.1), and platelets 92 K/µl (range, 24–112). At t(3;12) detection, all four MDS patients (100%) assessed for dysplasia demonstrated dysplasia. One MDS-excess blasts-2 (MDS-EB2) patient (#2) was not assessed due to too few non-blast cells. The dysplastic features identified in this group are as follows: granulocytic precursors (irregular nuclear contours including pseudo-Pelger-Huet forms), erythroid precursors (irregular nuclear contours including nuclear budding, nuclear cytoplasmic asynchrony, megaloblastoid changes, and basophilic stippling), megakaryocytes (small, nuclear hypolobation, separated nuclear lobes, and with hypogranulated platelets). At t(3;12) detection, one t-MDS had myelodysplasia in all three lineages, while one t-MDS patient and two MDS-EB2 patients had myelodysplasia in two lineages. The findings are summarized in Table 1.
At the time t(3;12) was detected, three patients were classified as myeloproliferative neoplasms, two had chronic myeloid leukemia, BCR-ABL1-positive, blast phase (CML-BP), and one had primary myelofibrosis (PMF). In 1 CML-BP case (#7) and the PMF case, the t(3;12) was detected later in the disease course. The first patient, after 22 months of CML, BCR-ABL1-positive, chronic phase (CML-CP) presented to our institution with 53% myeloid blasts in the blood, 85% hypercellular BM with 58% myeloid blasts in the aspirate and myelofibrosis (MF-2). The second CML-BP patient initially had CML-CP for 1 year and transformed to blast phase with 16% blasts in blood, 30% cellular marrow with 27% blasts in the aspirate and myelofibrosis (MF-2). One patient with PMF presented with 80% hypercellular BM with megakaryocytic hyperplasia with occasional large forms, no increase in blasts, and elevated platelets of 917 K/µl. Of the three MPN patients, two were assessed for dysplasia at t(3;12) detection (case #1 and 5). The PMF patient (#5) demonstrated dysplasia. CML-BP patient (#1) did not demonstrate dysplasia and the CML-BP patient (#7) was not assessed for dysplasia due to too few non-blast cells. The dysplastic features identified in this group are as follows: granulocytic precursors (cytoplasmic hypogranularity and pseudo-Pelger-Huet forms) and megakaryocytes (small with nuclear hypolobation). At t(3;12) detection, one PMF had myelodysplasia in two lineages. The findings are summarized in Table 1. A predominance of monolobate or bilobate megakaryocytes as seen in myeloid neoplasms with inv(3) was not present in this cohort of patients with myeloid neoplasms with t(3;12).
Cytogenetic and molecular findings
Results of conventional cytogenetic analysis identified the t(3;12)(q26.2;p13) in all 20 patients (Table 2 and Fig. 2a). Seven (35%) patients had t(3;12)(q26.2;p13) as the sole chromosomal abnormality, and seven (35%) had two cytogenetic abnormalities, and the remaining six (30%) patients had complex karyotype (≥3 cytogenetic abnormalities). Apart from t(3;12), chromosome 7 abnormalities were observed in eight patients including monosomy 7 (n = 5). One patient (5%) each had add(7q11.2), del(7q11.2), and del(7q22q36). Chromosome 22 abnormalities were observed in five (25%) patients including three with t(9;22) as well as monosomy 22 or idic(22)(p11.2) observed in one patient (5%) each.

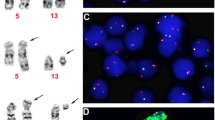
a Conventional study revealed: 46,XX,t(3;12)(q26.2;p13). b FISH using ETV6 dual-color break-apart probe revealed split signals, indicating ETV6 rearrangement. c FISH using MECOM dual-color break-apart probe revealed split signals, indicating MECOM rearrangement.
FISH studies were performed to assess MECOM in 11 patients and ETV6 in 7 patients. Rearrangements of MECOM and ETV6 were detected in all tested patients (Fig. 2b, c). Due to an obvious t(3;12) abnormality observed in almost all of analyzed metaphases (90–100%) by chromosomal analysis in five cases, no MECOM or ETV6 FISH were performed.
The de novo group consisted of the nine patients who had t(3;12) at time of initial diagnosis. This included five AML (#3, 8, 10, 11, and 19), three MDS-EB2 (#2, 9, and 14), and one had CML-BP (#1). Eleven patients acquired t(3;12) a median of 16.2 months (range, 7.2–60.8) after initial diagnosis (secondary group). This group included six AML (#4, 12, 15–17, and 20), two t-MDS (#6 and 13), one CML-BP (#7), one PMF (#5), and one MPAL (#18).
The de novo AML group (n = 5) had 11 genetic abnormalities (BCOR, ETNK1, FLT3 ITD x2, FLT3 exon 15, KIT, KRAS, NRAS x2, PTPN11, and RUNX1). The AML group where the t(3;12) was detected later in the disease course (n = 6) plus the one MPAL patient had 20 genetic abnormalities (BCR-ABL1, BRAF, DNMT3A, FLT3 ITD x2, FLT3 TKD, GATA2, IKZF1, KIT, KRAS x2, NRAS x2, PTPN11 x2, RUNX1, SF3B1 x2, U2AF1, and WT1). The variant allele frequency (VAF) for the former may have a lower trend, median 8.0% (range 7–46%, n = 5), compared to the secondary disease group, median 21.0% (range 8–47%, n = 9), but was not significantly lower (p = 0.21). VAFs reported as “<5%” and mutations without reported VAFs were not included in the calculations. The lack of significance differences in the two groups is likely due to the small sample sizes (Fig. 3).

a Distribution of mutations of myeloid neoplasms where t(3;12) was detected initially. b Distribution of mutations of myeloid neoplasms where t(3;12) was detected later in disease course. Each column represents a unique patient with myeloid neoplasm associated with t(3;12)(q26.2;p13) (n = 20). Each row represents a mutated gene or gene fusion identified in at least one patient. Numbers, when present, indicate percentage of variant allele frequency (VAF). Red empty boxes when VAF was unavailable. % = percentage of bone marrow blasts at detection of t(3;12); ND = percentage of blasts at detection of t(3;12) not determined.
The de novo MDS group (n = 3) had one genetic abnormality (SF3B1), while the secondary t-MDS group (n = 2) where the t(3;12) was detected later in the disease course had three genetic abnormalities (KRAS, NRAS, and TET2). The VAF for former group with the SF3B1 mutation was 10%, while for the secondary t-MDS group is median 11.5% (range 7–16%, n = 2). VAFs reported as “<5%” were not included in the calculations.
Treatment, response, and prognosis
Nineteen patients had treatment data available. All 19 received chemotherapy, including 14 who received hypomethylating drugs such as decitabine, guadecitabine, or azacitidine; 12 received antimetabolite chemotherapeutic agents such as cytarabine, fludarabine, or clofarabine; 11 (55%) received topoisomerase II inhibitors such as idarubicin, daunorubicin, mitoxantrone, or etoposide; 4 (20%) received immunotherapies such as anti-CD20, anti-CD33, anti-CD45, or anti-CD123; 4 (20%) patients received venetoclax or bortezomib; 3 (15%) received the FLT3: inhibitors sorafenib or gilteritinib; 2 (10%) received BCR-ABL1 tyrosine kinase inhibitors imatinib or dasatinib; and 1 (5%) patient was treated with anagrelide, methotrexate, pembrolizumab, revlimid, or vincristine. Nine (45%) patients underwent SCT: seven allogeneic and two autologous (Table 1). The autologous SCTs were for a patient with DLBCL and one patient with PCM, both of whom later developed therapy-related myeloid neoplasms.
Seven patients relapsed (#1, 3, 10, 15, and 17–19), defined as recurrence of the original disease. Five patients received allogeneic SCT and relapsed after a median duration of 11 months (range 1–28). Two patients (#6 and 16) received autologous SCT for nonmyeloid-related neoplasms before developing t-MDS. Patient (#6) received an autologous SCT for DLBCL, received an allogeneic SCT, and developed MDS then AML 11.1 and 31.2 months after the transplant, respectively. Patient (#15) received two allogeneic SCTs and relapsed 28 and 5 months after the first and second transplants, respectively. Two patients with allogeneic SCT remained in remission after 79.3 and 22 months of follow-up, respectively. Ten patients never achieved complete remission (#2, 4–8, 11, 12, 16 and 20). Four patients were lost to follow-up (two after allogeneic SCT, one after complete response, and one after relapse).
Clinical follow-up was available for all 20 patients. The median follow-up after detection of t(3;12) was 6.6 months (range, 0.1–150). During follow-up, ten patients had persistent disease and seven relapsed. Four patients with MDS (#4, 16, 17, and 20) progressed to AML after a median of 9.7 months (range, 6.2–19.5). Of the 14 patients who died by the end of the study, the median survival was 6.3 months (range, 0.1–17.3) after detection of t(3;12).
Discussion
The t(3;12) rearrangement is a rare event in hematologic neoplasms. In a study of 9550 cases of hematologic neoplasms with cytogenetic data, only ten (0.1%) had t(3;12) [13, 19]. Here we report 20 patients with hematologic neoplasms associated with t(3;12)(q26.2;p13). Fifteen cases were tested by FISH for MECOM (n = 11) and/or ETV6 (n = 7) rearrangements. All patients in this study group had myeloid neoplasms including AML (n = 11), MDS (n = 5), CML-BP (n = 2), PMF (n = 1), and MPAL (n = 1). In our experience at a large cancer center, we identified no cases of t(3;12) associated with lymphoid neoplasms, which is consistent with the findings in the literature.
A total of 58 reports of t(3;12)(q26.2;p13) have been published in the English literature as case reports, case series, or as karyotypes in larger studies (Tables 3 and 4). There were 34 men and 23 women (gender not reported for one patient) with a median age of 47.5 years (range, 4.8–87). We reviewed these published cases and reclassified the neoplasms, when possible, according to the WHO 2017 [42]. All cases reported in the literature have been myeloid neoplasms. AML was most common with 43 (74%) cases, followed by MDS with 9 (16%) cases, and 6 CML cases (10%, 3 CML-BP, 2 CML-CP, and 1 atypical CML, BCR-ABL1-negative).
In our study group, the t(3;12)(q26.2;p13) was present at time of initial diagnosis in 9 (45%) patients and acquired during the course of disease in the other 11 (55%) patients. This observation suggests that t(3;12) is a driver that can be an initiating event or occurs as a secondary event in disease progression. Seven (35%) patients had t(3;12)(q26.2;p13) as the sole chromosomal abnormality, while 13 (65%) patients had either 2 or ≥3 chromosomal abnormalities. Similar to our experience, cases with t(3;12)(q26.2;p13) reported in the literature also frequently had this translocation as a sole chromosomal abnormality (n = 29, 50%), while 21 (36%) had 2 chromosomal abnormalities, and 8 (14%) had ≥3 chromosomal abnormalities. However, the over representation of the proportion of our patients (30%) with ≥3 chromosomal abnormalities as compared to the literature (14%) may reflect a greater percentage of patients presenting to our institution who had already received multiple prior therapies.
The 29 cases in the literature with t(3;12) as the sole cytogenetic abnormality at initial diagnosis included 21 AML and 6 MDS (2 are MDS-EB) and 1 MDS/MPN-RS-T. In addition to t(3;12), monosomy 7 was the most common chromosomal abnormality reported both in literature (n = 8, 14%) and found in our study (n = 5, 25%) [19]. Cytogenetic abnormalities in addition to t(3;12) such as abnormalities of 7q and chromosome 22 did not significantly differ from the literature. It is important to note that the underrepresentation of other cytogenetic abnormalities reported in the literature may be due to the limited number of cases in this study.
t(3;12)(q26.2;p13) is also associated with a resistance to conventional chemotherapy and it confers a poorer prognosis. Of the 20 patients with follow-up information available, 10 patients had persistent disease, while other 7 patients relapsed. Of the 14 (70%) patients that died, the median survival was 6.3 months (range, 0.1–17.3 months) after detection of t(3;12)(q26.2;p13).
The literature reports MECOM abnormalities to be a poor prognostic indicator for AML patients [15]. Inappropriate overexpression of MECOM can occur via the t(3;12)(q26.2;p13) resulting in poorer prognosis [13]. Similar to our results, Kim et al. reported that the t(3;12)(q26.2;p13) in myeloid neoplasms results in leukemogenesis by promoting proliferative capacity and suggests a dismal prognosis, measured in a few months [13, 22, 40]. Lugthart et al. reported that patients with t(3;12)(q26.2;p13) showed 5-year survival rates of 25–51%. There are reports of patients with t(3;12)(q26.2;p13) that did not achieve complete remission with therapy and died of refractory disease [27, 33, 38]. Reports suggest that this translocation is associated with CML-BP, transformation of MDS or chronic myelomonocytic leukemia to AML, and a poor prognosis [12, 32]. One study of four patients had a median survival of 9.5 months (range, 2–12) [30]. Treatment failure in patients with AML associated with t(3;12)(q26.2;p13) may also be due to the fact that AML is secondary to MDS with the presence of additional chromosome 7 abnormalities [33]. Similar to the cases we report, the literature reports failure of therapy including patients who have undergone allogeneic SCT transplantation [30, 43].
Morphologic dysplasia was found in 11 of 12 (90%) cases assessed. Furthermore, either MDS defining cytogenetic abnormalities or dysplasia was found in 18 of 20 (90%) cases in our cohort as case #2, 8, and 12 demonstrated dysplasia after the detection of t(3;12). When comparing the subgroups of myeloid neoplasms, morphologic dysplasia was found in all the AML cases (5/5), MDS cases (4/4), half the myeloproliferative neoplasms cases (1/2), and one MPAL case assessed. All of the therapy-related cases (3/3) showed dysplasia.
Of the 58 cases described in the literature to date, 15 (26%) had MDS defining cytogenetic abnormalities. Of the 14 cases in the literature assessed for morphologic dysplasia, 14 cases (100%) demonstrated dysplasia. This is similar to the dysplasia found in the 90% of cases (11 of 12 cases assessed) in our study. Six (10.3%) of the 58 cases had both MDS defining cytogenetic abnormalities and dysplasia.
The role of t(3;12) in leukemogenesis is believed to result from the active promoter ETV6 inducing overexpression of EVI1 [12, 44]. Ottema et al. demonstrated that cases of inv(3) and t(3;3) exhibited EVI1 overexpression but low or no MDS1-EVI1 transcription (atypical 3q26-rearranged AML) due to translocation of the GATA2 enhancer to the MECOM locus. Given the clinical and biological similarities, the authors conclude that (1) atypical 3q26-rearranged AMLs and AMLs with inv(3)/t(3;3) are one entity and (2) routine analysis determining MECOM rearrangements require MECOM FISH to asses for MECOM rearrangement together with quantitative assessment of the EVI1 and MDS1-EVI1 mRNA expression levels [11]. In the future, it would be interesting to assess the EVI1 expression levels to see if t(3;12) has a similar mechanism of leukemogenesis as 3q26-rearranged AMLs. MECOM acts as a transcription factor that targets genes directly through its proximal zinc finger domain [45]. MECOM regulates the expression of GATA2, Pbx homeobox 1 (Pbx1), PROMYELOCYTE LEUKEMIA (PML) gene transcription, and inhibits the transforming growth factor beta (TGF-β) signaling by repressing mothers against decapentaplegic homolog 3 function by interacting with and recruiting corepressor C-terminal-binding protein [46,47,48,49,50]. Experimental evidence suggests that the leukemogenic effects also include promoting cell cycle proliferation by upregulating activator protein 1, blocking the growth inhibitory effects of TGF-β, cooperating with RAS/receptor tyrosine kinase pathway activating mutations, and suppressing apoptosis/differentiation [44, 51,52,53].
In summary, the presence of t(3;12)(q26.2;p13) is a rare abnormality associated exclusively with myeloid neoplasms. Morphologic myelodysplasia was found in all the AML cases (5/5), MDS cases (4/4), therapy-related cases (3/3), half the myeloproliferative neoplasms cases (1/2), and one MPAL case assessed. This translocation is associated with resistance to treatment, rapid disease progression, and a poorer prognosis. Novel therapeutic strategies are needed for this subset of patients with myeloid neoplasms. Given the close relationship between the presence of this cytogenetic abnormality and MDS-related changes, we recommend adding t(3;12)(q26.2;p13) to the list of AML-MRC-defining abnormalities of WHO 2017 classification of myeloid neoplasms.
References
Morishita K, Parker DS, Mucenski ML, Jenkins NA, Copeland NG, Ihle JN. Retroviral activation of a novel gene encoding a zinc finger protein in IL-3-dependent myeloid leukemia cell lines. Cell. 1988;54:831–40.
Bernstein R, Pinto MR, Behr A, Mendelow B. Chromosome 3 abnormalities in acute nonlymphocytic leukemia (ANLL) with abnormal thrombopoiesis: report of three patients with a “new” inversion anomaly and a further case of homologous translocation. Blood. 1982;60:613–7.
Bitter MA, Neilly ME, Le Beau MM, Pearson MG, Rowley JD. Rearrangements of chromosome 3 involving bands 3q21 and 3q26 are associated with normal or elevated platelet counts in acute nonlymphocytic leukemia. Blood. 1985;66:1362–70.
Carbonell F, Hoelzer D, Thiel E, Bartl R. Ph1-positive CML associated with megakaryocytic hyperplasia and thrombocythemia and an abnormality of chromosome no. 3. Cancer Genet Cytogenet. 1982;6:153–61.
Jotterand Bellomo M, Parlier V, Muhlematter D, Grob JP, Beris P. Three new cases of chromosome 3 rearrangement in bands q21 and q26 with abnormal thrombopoiesis bring further evidence to the existence of a 3q21q26 syndrome. Cancer Genet Cytogenet. 1992;59:138–60.
Pintado T, Ferro MT, San Roman C, Mayayo M, Larana JG. Clinical correlations of the 3q21;q26 cytogenetic anomaly. A leukemic or myelodysplastic syndrome with preserved or increased platelet production and lack of response to cytotoxic drug therapy. Cancer. 1985;55:535–41.
Nucifora G. The EVI1 gene in myeloid leukemia. Leukemia. 1997;11:2022–31.
Storlazzi CT, Anelli L, Albano F, Zagaria A, Ventura M, Rocchi M, et al. A novel chromosomal translocation t(3;7)(q26;q21) in myeloid leukemia resulting in overexpression of EVI1. Ann Hematol. 2004;83:78–83.
Morishita K, Parganas E, William CL, Whittaker MH, Drabkin H, Oval J, et al. Activation of EVI1 gene expression in human acute myelogenous leukemias by translocations spanning 300-400 kilobases on chromosome band 3q26. Proc Natl Acad Sci USA. 1992;89:3937–41.
De Weer A, Speleman F, Cauwelier B, Van Roy N, Yigit N, Verhasselt B, et al. EVI1 overexpression in t(3;17) positive myeloid malignancies results from juxtaposition of EVI1 to the MSI2 locus at 17q22. Haematologica. 2008;93:1903–7.
Ottema S, Mulet-Lazaro R, Beverloo HB, Erpelinck CAJ, van Herk S, Helm RV, et al. Atypical 3q26/MECOM rearrangements genocopy inv(3)/t(3;3) in acute myeloid leukemia. Blood. 2020;136:224–34.
Peeters P, Wlodarska I, Baens M, Criel A, Selleslag D, Hagemeijer A, et al. Fusion of ETV6 to MDS1/EVI1 as a result of t(3;12)(q26;p13) in myeloproliferative disorders. Cancer Res. 1997;57:564–9.
Achkar WA, Aljapawe A, Liehr T, Wafa A. De novo acute myeloid leukemia subtype-M4 with initial trisomy 8 and later acquired t(3;12)(q26;p12) leading to ETV6/MDS1/EVI1 fusion transcript expression: a case report. Oncol Lett. 2014;7:787–90.
Bacher U, Kern W, Schnittger S, Hiddemann W, Schoch C, Haferlach T. Blast count and cytogenetics correlate and are useful parameters for the evaluation of different phases in chronic myeloid leukemia. Leuk Lymphoma. 2005;46:357–66.
Barjesteh van Waalwijk van Doorn-Khosrovani S, Erpelinck C, van Putten WL, Valk PJ, van der Poel-van de Luytgaarde S, Hack R, et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood. 2003;101:837–45.
Beyer V, Castagne C, Muhlematter D, Parlier V, Gmur J, Hess U, et al. Systematic screening at diagnosis of -5/del(5)(q31), -7, or chromosome 8 aneuploidy by interphase fluorescence in situ hybridization in 110 acute myelocytic leukemia and high-risk myelodysplastic syndrome patients: concordances and discrepancies with conventional cytogenetics. Cancer Genet Cytogenet. 2004;152:29–41.
Bobadilla D, Enriquez EL, Alvarez G, Gaytan P, Smith D, Slovak ML. An interphase fluorescence in situ hybridisation assay for the detection of 3q26.2/EVI1 rearrangements in myeloid malignancies. Br J Haematol. 2007;136:806–13.
Dastugue N, Lafage-Pochitaloff M, Pages MP, Radford I, Bastard C, Talmant P, et al. Cytogenetic profile of childhood and adult megakaryoblastic leukemia (M7): a study of the Groupe Francais de Cytogenetique Hematologique (GFCH). Blood. 2002;100:618–26.
Haferlach C, Bacher U, Schnittger S, Alpermann T, Zenger M, Kern W, et al. ETV6 rearrangements are recurrent in myeloid malignancies and are frequently associated with other genetic events. Genes Chromosomes Cancer. 2012;51:328–37.
Hazard E, Bastard C, Callat MP, Schneider P, Vannier JP. De novo acute myeloblastic leukemia with trilineage abnormalities and a t(3;12) in a child. Med Pediatr Oncol. 2003;40:397–400.
Iwase S, Furukawa Y, Horiguchi-Yamada J, Nemoto T, Takahara S, Kawano T, et al. A novel variant of acute myelomonocytic leukemia carrying t(3;12)(q26;p13) with characteristics of 3q21q26 syndrome. Int J Hematol. 1998;67:361–8.
Kim YJ, Yang JJ, Han Y, Kim S, Yang HS, Yoon HJ, et al. A rare case of ETV6/MECOM rearrangement in therapy-related acute myeloid leukemia with t(3;12) and monosomy 7. Clin Lab. 2017;63:415–8.
Langabeer SE, Rogers JR, Harrison G, Wheatley K, Walker H, Bain BJ, et al. EVI1 expression in acute myeloid leukaemia. Brit J Haematol. 2001;112:208–11.
Lavallee VP, Gendron P, Lemieux S, D’Angelo G, Hebert J, Sauvageau G. EVI1-rearranged acute myeloid leukemias are characterized by distinct molecular alterations. Blood. 2015;125:140–3.
Lugthart S, Groschel S, Beverloo HB, Kayser S, Valk PJ, van Zelderen-Bhola SL, et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol. 2010;28:3890–8.
Nakamura Y, Nakazato H, Sato Y, Furusawa S, Mitani K. Expression of the TEL/EVI1 fusion transcript in a patient with chronic myelogenous leukemia with t(3;12)(q26;p13). Am J Hematol. 2002;69:80–2.
Nishimura Y, Wada H, Mori A, Takatsuka H, Tamura A, Fujimori Y, et al. Detection of ETV6/MDS1/Evi-1 chimeric transcripts in a patient with acute myelocytic leukemia and t(3;12)(q26;p13). Int J Hematol. 2000;72:108–9.
Paquette RL, Nicoll J, Chalukya M, Elashoff D, Shah NP, Sawyers C, et al. Frequent EVI1 translocations in myeloid blast crisis CML that evolves through tyrosine kinase inhibitors. Cancer Genet. 2011;204:392–7.
Poppe B, Dastugue N, Vandesompele J, Cauwelier B, De Smet B, Yigit N, et al. EVI1 is consistently expressed as principal transcript in common and rare recurrent 3q26 rearrangements. Gene Chromosomes Cancer. 2006;45:349–56.
Raynaud SD, Baens M, Grosgeorge J, Rodgers K, Reid CD, Dainton M, et al. Fluorescence in situ hybridization analysis of t(3; 12)(q26; p13): a recurring chromosomal abnormality involving the TEL gene (ETV6) in myelodysplastic syndromes. Blood. 1996;88:682–9.
Roumier C, Eclache V, Imbert M, Davi F, MacIntyre E, Garand R, et al. M0 AML, clinical and biologic features of the disease, including AML1 gene mutations: a report of 59 cases by the Groupe Francais d’Hematologie Cellulaire (GFHC) and the Groupe Francais de Cytogenetique Hematologique (GFCH). Blood. 2003;101:1277–83.
Secker-Walker LM, Mehta A, Bain B. Abnormalities of 3q21 and 3q26 in myeloid malignancy: a United Kingdom Cancer Cytogenetic Group study. Br J Haematol. 1995;91:490–501.
Voutsadakis IA, Maillard N. Acute myelogenous leukemia with the t(3;12)(q26;p13) translocation: case report and review of the literature. Am J Hematol. 2003;72:135–7.
Yamamoto K, Yakushijin K, Funakoshi Y, Inui Y, Okamura A, Matsuoka H, et al. Biallelic ETV6 rearrangements by recurrent translocations t(7;12)(p15;p13) and t(3;12)(q26.2;p13) in acute myeloid leukemia. Leuk Res. 2011;35:e212–4.
Orciuolo E, Buda G, Cecconi N, Galimberti S, Petrini M. A therapy resistant myelodysplastic syndrome characterized by the presence of the rare reciprocal translocation t(3;12)(q26.2;p13). Leuk Res. 2007;31:1599–600.
Shimada K, Tomita A, Minami Y, Abe A, Hind CK, Kiyoi H, et al. CML cells expressing the TEL/MDS1/EVI1 fusion are resistant to imatinib-induced apoptosis through inhibition of BAD, but are resensitized with ABT-737. Exp Hematol. 2012;40:724–37.e2.
van de Loosdrecht AA, Kok K, de Vries B, van der Veen AY, van den Berg E, Esselink MT, et al. Breakpoint analysis by fluorescence in situ hybridization in myelodysplastic syndromes with t(3;12)(q26;p13) and expression of EVI1. Leukemia. 2000;14:1857–8.
Taylor G, Culligan D, Vickers MA. Refractory anemia with ring sideroblasts associated with marked thrombocytosis complicated by massive splenomegaly treated with lenalidomide resulting in resolution of splenomegaly but severe and prolonged pancytopenia. Case Rep Hematol. 2013;2013:718480.
Yamamoto K, Yakushijin K, Ichikawa H, Okamura A, Nagao S, Kakiuchi S, et al. Coexpression of ETV6/MDS1/EVI1 and ETV6/EVI1 fusion transcripts in acute myeloid leukemia with t(3;12)(q26.2;p13) and thrombocytosis. Leuk Lymphoma. 2019;60:1294–8.
Wang XI, Xinyan L, Yin C, Zhao L, Bueso-Ramos CE, Medeiros LJ, et al. Myeloid neoplasms associated with t(3;12)(q26.2;P13) are clinically aggressive and frequently harbor FLT3 mutations: a report of 8 cases and review of literature. J Leuk. 2014;2:1–8.
Wang W, Routbort MJ, Tang Z, Ok CY, Patel KP, Daver N, et al. Characterization of TP53 mutations in low-grade myelodysplastic syndromes and myelodysplastic syndromes with a non-complex karyotype. Eur J Haematol. 2017;99:536–43.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Massaad L, Prieur M, Leonard C, Dutrillaux B. Biclonal chromosome evolution of chronic myelomonocytic leukemia in a child. Cancer Genet Cytogen. 1990;44:131–7.
De Braekeleer M, Le Bris MJ, De Braekeleer E, Basinko A, Morel F, Douet-Guilbert N. 3q26/EVI1 rearrangements in myeloid hemopathies: a cytogenetic review. Future Oncol. 2015;11:1675–86.
Yatsula B, Lin S, Read AJ, Poholek A, Yates K, Yue D, et al. Identification of binding sites of EVI1 in mammalian cells. J Biol Chem. 2005;280:30712–22.
Yuasa H, Oike Y, Iwama A, Nishikata I, Sugiyama D, Perkins A, et al. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005;24:1976–87.
Shimabe M, Goyama S, Watanabe-Okochi N, Yoshimi A, Ichikawa M, Imai Y, et al. Pbx1 is a downstream target of Evi-1 in hematopoietic stem/progenitors and leukemic cells. Oncogene. 2009;28:4364–74.
Buonamici S, Li D, Mikhail FM, Sassano A, Platanias LC, Colamonici O, et al. EVI1 abrogates interferon-alpha response by selectively blocking PML induction. J Biol Chem. 2005;280:428–36.
Izutsu K, Kurokawa M, Imai Y, Maki K, Mitani K, Hirai H. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor beta signaling. Blood. 2001;97:2815–22.
Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, et al. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature. 1998;394:92–6.
Glass C, Wilson M, Gonzalez R, Zhang Y, Perkins AS. The role of EVI1 in myeloid malignancies. Blood Cell Mol Dis. 2014;53:67–76.
Groschel S, Sanders MA, Hoogenboezem R, Zeilemaker A, Havermans M, Erpelinck C, et al. Mutational spectrum of myeloid malignancies with inv(3)/t(3;3) reveals a predominant involvement of RAS/RTK signaling pathways. Blood. 2015;125:133–9.
Metais JY, Dunbar CE. The MDS1-EVI1 gene complex as a retrovirus integration site: impact on behavior of hematopoietic cells and implications for gene therapy. Mol Ther. 2008;16:439–49.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ronaghy, A., Hu, S., Tang, Z. et al. Myeloid neoplasms associated with t(3;12)(q26.2;p13) are clinically aggressive, show myelodysplasia, and frequently harbor chromosome 7 abnormalities. Mod Pathol 34, 300–313 (2021). https://doi.org/10.1038/s41379-020-00663-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00663-z
This article is cited by
-
Antineoplastics
Reactions Weekly (2021)
