
Abstract
Childhood B-cell acute lymphoblastic leukaemia (B-ALL) is characterised by recurrent genetic abnormalities that drive risk-directed treatment strategies. Using current techniques, accurate detection of such aberrations can be challenging, due to the rapidly expanding list of key genetic abnormalities. Whole genome sequencing (WGS) has the potential to improve genetic testing, but requires comprehensive validation. We performed WGS on 210 childhood B-ALL samples annotated with clinical and genetic data. We devised a molecular classification system to subtype these patients based on identification of key genetic changes in tumour-normal and tumour-only analyses. This approach detected 294 subtype-defining genetic abnormalities in 96% (202/210) patients. Novel genetic variants, including fusions involving genes in the MAP kinase pathway, were identified. WGS results were concordant with standard-of-care methods and whole transcriptome sequencing (WTS). We expanded the catalogue of genetic profiles that reliably classify PAX5alt and ETV6::RUNX1-like subtypes. Our novel bioinformatic pipeline improved detection of DUX4 rearrangements (DUX4-r): a good-risk B-ALL subtype with high survival rates. Overall, we have validated that WGS provides a standalone, reliable genetic test to detect all subtype-defining genetic abnormalities in B-ALL, accurately classifying patients for the risk-directed treatment stratification, while simultaneously performing as a research tool to identify novel disease biomarkers.
Similar content being viewed by others


Introduction
Childhood and adolescent B-cell acute lymphoblastic leukaemia (B-ALL) is one of the success stories of modern medicine, with survival rates exceeding 90% [1]. Risk stratification for treatment within contemporary clinical trials has contributed significantly to this achievement, through classification into risk groups based on genetic subtyping and minimal residual disease (MRD) assessment [2]. Approximately 70% of childhood B-ALL are currently routinely characterised by established cytogenetic abnormalities, associated with good or poor outcomes [3]. The remaining 30% of patients, lacking established chromosomal aberrations and termed “B-other-ALL”, were collectively assigned to the intermediate risk group. More recently, genomic approaches have identified new genetic subtypes among B-other-ALL, including DUX4-rearranged (DUX4-r, mostly IGH::DUX4), ABL-class fusions, and MEF2D-rearranged (MEF2D-r), which have been associated with specific clinical characteristics and differing outcomes (Table 1) [4,5,6,7,8,9,10]. Thus, their accurate detection is increasingly important for assignment to the most appropriate therapy, as now well-established for ABL-class fusions and treatment with tyrosine kinase inhibitor (TKI) therapy [11, 12].
A range of standard-of-care techniques are currently used to detect clinically-relevant abnormalities in B-ALL, including karyotyping, fluorescence in situ hybridisation (FISH) and SNP arrays [13, 14]. As the list of subtype-defining genetic abnormalities has grown, multiple tests are often required. In addition, it has been difficult to achieve a single test for some important abnormalities, such as DUX4-r, which defines a distinct subtype of B-ALL with a good prognosis [6, 7]. Detection of this rearrangement is challenging due to the repetitive and variable sequences involved [15]. Molecular schema for B-ALL subtyping using whole transcriptome sequencing (WTS) have recently been described, but they require large reference datasets as they are focused on global expression profiles [7, 16]. These challenges, together with the lack of recurrent genetic abnormalities in around 10% of patients [17], drive the need for enhanced genetic diagnostic tools in B-ALL.
Genetic testing using whole genome sequencing (WGS) is becoming increasingly feasible, as many of the analytical demands are being addressed [18, 19]. Indeed, some national healthcare providers, such as the NHS Genomic Medicine Service and Genomic Medicine, Sweden, are offering WGS as a genetic test for haematological and other paediatric malignancies [20]. To date, WGS has been used to explore the genomic landscape of B-ALL subtyped by WTS [17]. A DNA-based molecular schema for subtyping with comprehensive validation of WGS as a standalone diagnostic tool in B-ALL has not been performed. In this study of a large cohort of genetically and clinically well-annotated childhood and adolescent B-ALL, we have produced such a schema for accurate subtyping by WGS, which has clearly demonstrated the future role of WGS for improved genetic risk-directed stratification compared to previous standard-of-care approaches.
Materials and methods
Patient information
Diagnostic bone marrow samples from 210 childhood B-ALL were included in this study, and divided into two cohorts: (1) known cytogenetic abnormalities of clinical significance (n = 38) (patients 1–38); (2) no established cytogenetic abnormalities detected by standard-of-care methods and treated on the UK childhood ALL treatment trial, UKALL2003 (defined as “B-other-ALL” at the time of the trial, n = 172) (patients 39–210, Supplementary Table 1, Supplementary Information) [21, 22]. All patients were diagnosed using standard morphological and immunophenotyping methods. Leukaemic blast count at diagnosis was ≥70% in 92% (163/177) of patients with information available. Post-treatment bone marrow samples were used as matched germline controls for 208 patients (Supplementary Table 1, Supplementary Information).
Whole genome sequencing (WGS)
WGS was performed on 210 diagnostic (mean sequencing depth of 76×) and 208 matched remission (mean sequencing depth of 38×) DNA samples, as described in Supplementary Information.
Reads were aligned to Human Reference genome version 38 (GRCh38), and germline and somatic variants were identified, as described in Supplementary Information. We considered structural variants (SV) or copy number variants/aberrations (CNV/CNA) that were located within or surrounding (≤10 kb) a gene, as well as single nucleotide variants (SNVs) and indels that were located within the coding sequence of a gene. Tumour-only (T-only) analysis was performed on all patients with established genetic abnormalities of clinical significance (n = 38, cohort 1) and diagnostic samples without a matched germline (n = 2), as described in Supplementary Information.
Individual B-ALL samples (n = 210) were classified into subtypes based on the detection of specific genetic features (Table 1, Supplementary Information). In cases with genetic alterations characteristic of ≥2 subtypes, the primary subtype was assigned as the one with the highest supporting read count, except for DUX4-r cases, due to difficulties in mapping reads to these regions. Cases lacking obvious defining features were classified as ‘other’.
Detection of DUX4-rearrangements by WGS
A customised T-only pipeline was developed for the detection of IGH::DUX4 spanning reads in our B-ALL samples (n = 210). Available matched germline samples (n = 208) were used to determine the baseline level of spanning reads expected to be false positive alignments. Spanning read pairs from all samples with >10 spanning reads per billion (SRPB) were locally assembled to generate contigs and scaffolds used for IGH::DUX4. Cases of DUX4-r with other (non-IGH) partner genes were identified, as described in Supplementary Information.
Detection of clinically relevant genetic abnormalities by WGS
Cytogenetics, FISH and Multiplex Ligation-dependent Probe Amplification (MLPA) (MRC Holland, The Netherlands) were performed, as previously described [23,24,25]. Abnormalities detected by these methods are provided in Supplementary Table 2. Copy number data for nine genes/regions targeted in the SALSA MLPA P335 (CDKN2A/B, PAX5, IKZF1, BTG1, EBF1, RB1, ETV6, PAR1 region) and P327 (ERG) kits were included. Detection of risk-stratifying genetic abnormalities by WGS required these key features: (1) ploidy and focal CNA required CNA and/or SV information and (2) gene fusion required evidence of a SV [26, 27].
Cross validation using whole transcriptome sequencing (WTS)
WTS was performed on RNA samples, extracted from diagnostic bone marrow using the RNeasy Extraction kit (Qiagen, Manchester, UK), from 85 patients within the same B-ALL cohort. Sequencing reads were processed and aligned to Human Reference Genome GRCh38. Molecular classification and subtyping was performed, as previously described (Supplementary Information) [6, 7, 28].
Results
Robust detection of established cytogenetic abnormalities by WGS
We have demonstrated that WGS can reliably detect the important risk-stratifying genetic abnormalities in B-ALL by investigating the 38 samples with known chromosomal abnormalities (cohort 1) (Supplementary Fig. 1A). The automated DRAGEN T-only pipeline called 37/38 of the primary genetic abnormalities without analysis of the associated germline sample, while automated T-N analysis identified 34/38 of them. Using either approach, the patterns of whole chromosome gain or loss in subtypes associated with aneuploidy (high hyperdiploidy, low hypodiploidy, near-haploidy) and whole chromosome copy number-neutral loss-of-heterozygosity (CN-LOH), identifying masked near-haploidy/low hypodiploidy, were consistent with FISH and cytogenetic analyses (Supplementary Table 2). In addition, the complex genomic profile of ALL with intrachromosomal amplification of chromosome 21 (iAMP21-ALL, n = 3), and in-frame gene fusions of KMT2A-r (n = 8), ETV6::RUNX1 (n = 5) and TCF3::PBX1 (n = 3), were consistently identified using T-N and T-only approaches.
T-N analysis failed to identify the expected fusion gene in four patients: one of three BCR::ABL1 and all three EBF1::PDGFRB (ABL-class subtype) cases (Supplementary Table 2). Two factors affected their detection by the T-N automated pipeline: (1) high levels of residual leukaemic blasts (high MRD) in the germline sample. Aligned sequencing reads showed evidence of the fusion gene in both leukaemia and matched germline samples in all four patients (Supplementary Fig. 1B–E). Although remission bone marrow was easily accessible for use as a matched germline control in this study, other germline samples, including skin biopsies, hair or nail extracts, may be better options. Crucially, automated T-only analysis identified EBF1::PDGFRB in all three patients. (2) Complex rearrangement near the breakpoint. The BCR::ABL1 rearrangement showed a second rearrangement (duplication) visible in the aligned reads close (418 bp) to the BCR::ABL1 breakpoint, which had been called as a separate event by automated analysis (Supplementary Fig. 2). Interestingly, this same duplication close to the BCR::ABL1 breakpoint was found in a second BCR::ABL1 patient, which may indicate a recurring event. Importantly, all 38 risk-stratifying genetic abnormalities were successfully detected by WGS.
Molecular classification and subtyping of B-other-ALL by WGS
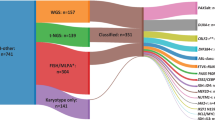
Next, we investigated the 172 cases in cohort 2 by WGS T-N analysis. Reinforcing the accuracy of WGS, 19 cases harboured established chromosomal abnormalities, which were previously undetected due to limited material and incomplete standard-of-care testing at the time of diagnosis: high hyperdiploidy (n = 5), iAMP21-ALL (n = 3), ETV6::RUNX1 (n = 1), TCF3::PBX1 (n = 8), TCF3::HLF (n = 1), low hypodiploidy (n = 1) (Supplementary Table 1, Supplementary Information, Fig. 1). The eight TCF3::PBX1 and one TCF3::HLF cases showed normal/undefined karyotypes and TCF3 FISH had not been performed. Among the remaining 153 cases, we were able to characterise 145 patients with cytogenetically-cryptic, subtype-defining genetic abnormalities (Supplementary Table 3, Fig. 2). The predominant subtype was DUX4-r (n = 59), followed by PAX5alt (n = 29), ZNF384-r (n = 12) and ETV6::RUNX1-like (n = 12). An additional DUX4-r, in association with iAMP21-ALL, was identified within cohort 1 (20724), bringing the total DUX4-r cases to 60. Two subtype-defining genetic abnormalities were identified in the same patient sample in eight cases, as indicated in Supplementary Table 3. Although the clinical significance of such co-existing abnormalities requires further assessment, their detection and estimate of subclonality was facilitated by WGS.

Oncoplot showing the subtype definition of each case and the associated subtype-defining genetic abnormalities observed by WGS. Colours define the subtype of each patient and the type of rearrangement for each genetic abnormality.

A Genetic subtypes as defined by WGS of 173 B-ALL patients, including 172 B-other-ALL patients (cohort 2) and one patient with iAMP21-ALL (cohort 1) in which a DUX4-r was observed. Eight patients had no subtype-defining genetic abnormalities (termed ‘other’). B t-distributed stochastic neighbour embedding (tSNE) plot of 85 B-other-ALL patients from this study (red triangles) and 1452 B-ALL patients from our previous study, demonstrating the subtype groupings (colour coded) of each patient based on WTS data [7]. This analysis validated the identification of recent subtype-defining genetic abnormalities that were identified within the WGS data.
Among the subtypes characterised by gene rearrangements, 55 different fusion genes were identified by WGS (Supplementary Table 4). These included three different partner genes of DUX4: IGH (n = 57) and novel partners MYB (n = 1, #23445) and DNTT (n = 1, #11148). One partner gene (#22355) remains unidentified, as discussed in Supplementary Information. PAX5 and ETV6 rearrangements were the most variable in relation to breakpoint and partner genes involved (Supplementary Fig. 3). Fusion genes involving PAX5 and ETV6 are characteristic of PAX5alt and ETV6::RUNX1-like ALL, respectively, which are often genetically complex (Supplementary Table 5) and associated with a range of underlying genetic abnormalities that drive global transcriptome profiles as defined by WTS [6, 29]. Here, WGS identified subtype-defining genetic abnormalities in all PAX5alt and ETV6::RUNX1-like cases, including a number of new aberrations. For example, a large insertion (>250 bp) involving exon 5 of PAX5 was observed in two PAX5alt cases, and five cases of PAX5alt were found to share a common profile of monoallelic PAX5 and biallelic CDKN2A/B losses, often with biallelic MTAP abnormalities (4/5 patients). Importantly, these cases lacked other subtype-defining genetic abnormalities and were validated as PAX5alt in those patients with matched WTS data (Fig. 3A). Separately, the mutational signature associated with the AID/APOBEC family of cytidine deaminases and a higher mutational load was demonstrated to be a robust associated genetic abnormality in ETV6::RUNX1-like patients, as previously reported (Fig. 3B, C) [17, 30]. Notably, the co-existence of an internal tandem duplication of IKZF1 (consistently involving exon 5) was observed in all IKZF1 N159Y patients in this study (3/3). The wide range of genetic profiles detected within B-other-ALL by WGS emphasises the challenges in their detection using standard-of-care techniques.

A Genetic abnormalities (CNA, SV, missense/frameshift/splice site mutations and small insertions (<500 bp)) involving individual exons of PAX5, CDKN2A, CDKN2B and MTAP in PAX5alt cases (n = 29). ‘CN profile’ describes a group of PAX5alt cases identified by WGS with PAX5 loss, biallelic CDKN2A and CDKN2B loss, often with MTAP abnormalities. PAX5alt subtyping was validated in all patients with matched WTS data (n = 17). B, C The mutational load in ETV6::RUNX1-like patients is shown to be elevated (median 2.91, range 0.63–6.5) (B), andthe AID/APOBEC family of cytidine deaminases represents >5% of the mutational signature profile in 10/12 ETV6::RUNX1-like cases (green) (C). Enrichment of the AID/APOBEC mutational signature is also evident in ETV6::RUNX1 patients (pink), as previously reported [30].
Characterisation of ‘other’ cases by WGS
Although no subtype-defining genetic abnormalities were observed in eight patients (Fig. 4), seven of them harboured genetic abnormalities that were clonal, recurrent in our cohort and/or located within known ALL-associated genes. A SH2B3 mutation in combination with gain of chromosome 21 was identified in one patient (n = 1, #10868), an association that we have previously reported [31]. Two patients had abnormalities of CNTNAP3B, a gene previously reported to be rearranged in infant KMT2A-r ALL cases: [32] a CNTNAP3B::C20orf203 fusion (#10868) and a missense mutation (Gly520Ala) (#22188). Multiple clonal rearrangements involving TCF3 and novel partner genes were identified in one patient (#23678), and a 1.5 MB (chr1:119983209-121429772) deletion was identified that targeted up to eight genes, including exons 1–6 of NOTCH2, in patient #24669. Previously unreported recurrent or clonal rearrangements involved genes within the mitogen-activated protein kinase (MAPK) pathway: UBA6_AS1::MAPK10 (n = 1, #21424) and RRAGB::MAPKAP1 (n = 1, #22188) (Supplementary Table 4).

The oncoplot provides details of genetic abnormalities that were clonal, recurrent or within ALL-associated genes detected by WGS. The primary subtype (black) defined by WTS is shown for four patients with matched WTS data. The subtype definition of each case based on Prediction Analysis of Microarrays (PAM) or two-dimensional t-distributed stochastic neighbour embedding (tSNE) analyses is shown. The presence of a subtype-defining fusion transcript in each sample is given; apart from patient 22980 with a ZNF618-NUTM1 fusion by WTS only. No fusion transcript was detected in the remaining patients.
We had matched WTS data for four of the above eight patients (Fig. 4). The results in relation to subtype definition by WGS concurred in three patients; the fourth case (22980) was classified as NUTM1-r by WTS alone. By WGS, no recurrent or clonal abnormalities were detected, even from inspection of aligned reads over NUTM1. However, it had a lower blast count (56% compared to an average blast count of 89% for the entire cohort), potentially explaining the inability to detect NUTM1-r, or any other clonal genetic feature, by WGS. As lower counts may hinder accurate detection of abnormalities, enrichment of blasts prior to DNA extraction or increased sequencing coverage in samples with <70% blasts, may be beneficial.
Improved detection and characterisation of DUX4 rearrangements
We developed a novel, customised analytical approach that identified 57 B-ALL patients with >10 spanning reads per billion (SRPB) between IGH and DUX4 (Fig. 5A, Supplementary Information, Supplementary Table 6). There was complete concordance in detection of DUX4-r among cases with both WTS and WGS (n = 21), including the case with DUX4::MYB and the patient with fewest supporting reads by WGS (SRPB, 11.1), demonstrating the accuracy of WGS in DUX4-r subtyping. Although all 21 DUX4-r cases had global transcriptome profiles associated with DUX4-r and overexpression of DUX4, an IGH::DUX4 fusion was only observed in 14/21 of the cases by WTS, while overlapping levels of DUX4 expression were found in non-DUX4-r cases (Supplementary Fig. 4). These discrepancies demonstrate the requirement for a comparator cohort and validated analyses before relying solely on WTS for accurate DUX4-r classification.

A Spanning reads per billion (SRPB) between the IGH and DUX4 loci. IGH::DUX4 patients (n = 57) were found to have 11.1-157.3 SRPB. ALL samples from other subtypes (and matched germline samples) show lower values, ranging from 0–9.6 SPRB. A threshold of >10 SRPB was applied to define patients with IGH::DUX4 abnormalities. Three DUX4-r patients did not show >10 SRPB as the rearrangement involved alternative genomic regions. B Breakpoint mapping of IGH::DUX4 breakpoint within the IGH locus of 53 patients. Breakpoints mapping to the forward (red) or reverse (brown) strand are shown. A cluster breakpoint region (CBR) within the IGH J (joining) segment is present, in which 47/53 cases harbour a breakpoint (chr14:105860602-105865246). C ERG abnormalities are seen in 68.3% (41/60) of DUX4-r patients. The exon structure of ERG is depicted in NM_001136154.1 and NM_182918.4 (not to scale); exons are numbered and represented with purple rectangles. The type of abnormalities range from deletion (pink), inversion (yellow), mutation (lollipop stick) and translocation (lollipop stick labelled ‘BND’). The width of the ribbon represents the number of cases with the abnormality.
It is estimated that 11-100 near-identical copies of DUX4 are repeated within the subtelomeric regions of chromosomes 4 and 10 [15]. To improve their characterisation, de novo assembly of sequencing reads in 53 IGH::DUX4 cases generated an average of 2.09 contigs/scaffolds per patient sample (Supplementary Table 7). The contigs/scaffolds revealed a common breakpoint region (CBR) within the IGH locus, chr14:105860602-105865246, in which 47/53 cases harboured a breakpoint (Fig. 5B). Interestingly, a second IGH breakpoint >100 kb distant from the first was observed in 24/53 of cases. Although 28% of DUX4-aligned sequences mapped to the chromosome 4 and 10 reference sequences with similar identity, 42% and 30% of DUX4-aligned sequences mapped more precisely to chromosome 4 or 10, respectively. In some cases, there was evidence of more complex rearrangement patterns, including IGH::DUX4::IGH to IGH::IGH::DUX4 (Supplementary Fig. 5). The transcriptional consequence of these complex rearrangement patterns could not be explored due to a lack of cases with matched WTS data.
Detection of focal abnormalities important for improved genetic classification of ALL
The UKALL-CNA classifier defines prognostic subtypes based on focal copy number changes in eight key genes/regions associated with B-ALL [33]. Furthermore, ERG deletions are considered to be a surrogate marker of DUX4-r due to them being found exclusively in this B-ALL subtype [10, 17, 34]. Somatic genetic abnormalities were detected in these key genes/regions in 172/208 patients from the WGS T-N cohort (Supplementary Table 8). The relative incidence and size of the focal genetic CNA varied between subtypes, as previously described (Supplementary Fig. 6, and Supplementary Table 9) [33, 35]. Among 367 genetic abnormalities observed by WGS, 332 involved CNA used in the UKALL-CNA classifier or within ERG, including seven focal deletions within PAR1 on chromosome X/Y, resulting in P2RY8::CRLF2 fusion. The remaining variants (n = 35) involved whole chromosome/chromosome arm gains, which are not included in the UKALL-CNA classifier.
Parallel WGS and MLPA data were available for 304/332 CNA observed by WGS within the UKALL-CNA classifier. Results were concordant for 242/304, while the remaining 62 CNA were called by WGS only (Supplementary Table 8). By MLPA, CNA were not called if: (1) the copy number level was outside the detection threshold (MLPA probe ratio <0.75 for deletions) (31/62 CNA) or (2) there was no or single MLPA probe coverage (≥two successive probes must be abnormal to call CNA by MLPA) (31/62 CNA) (Supplementary Fig. 7). These findings indicate that the recommended cut-off levels for positive MLPA results may be too stringent, as evidenced by the IKZF1 exonic duplications. They were exclusively observed in IKZF1 N159Y patients (n = 3), however they were not called by MLPA in two patients because the gains were restricted to a single probe (Supplementary Fig. 7B).
In this study, ERG abnormalities were observed in DUX4-r patients at an incidence of 68% (41/60) by WGS, compared to 35% (21/60) by MLPA. Most patients harboured a single ERG abnormality, but two distinct genetic variants were identified in five patients. The range of ERG abnormalities included deletion (n = 34), mutation (n = 8), inversion (n = 3) and translocation (n = 1) (Fig. 5C). Importantly, copy number profiling methods will only detect ERG deletions, while they will miss the small variants and translocations detectable by WGS.
Discussion
In this study, we have shown the excellent performance of WGS as a standalone diagnostic genetic screen in childhood B-ALL. T-only analysis provided rapid and accurate detection of those clinically-important genetic abnormalities required for risk stratification for treatment in a greater number of cases than standard-of-care techniques. In particular, we showed that WGS was most effective for the detection of the rapidly increasing list of newly reported and cytogenetically-cryptic abnormalities [7, 33, 36]. Combining T-only with T-N analysis, using appropriate germline samples and accepted mean sequencing depths provides fully comprehensive analysis of somatic variants for discovery of novel abnormalities and a deeper understanding of associated genetic changes.
Molecular subtypes defined by a range of genetic abnormalities present a challenge for accurate classification by current standard-of-care diagnostic tests. For example, PAX5alt and ETV6::RUNX1-like patients have unique gene expression profiles, but the driving genetic abnormalities are diverse and often undefined by WTS [5, 6]. Here, we have shown the significant contribution made by WGS in defining the complex genomic landscape underlying the PAX5alt and ETV6::RUNX1-like subtypes, highlighting some advantages of WGS over WTS, including detection of focal CNA and other rearrangements not involving fusion genes. Robust detection of all subtype-defining genetic abnormalities is key to future improvements in risk-directed therapy. Thus, the development of a DNA-based molecular schema that has been validated for accurate B-ALL subtyping using WGS in this way is timely.
In this study, we reported MAPK pathway gene fusions and CNTNAP3B abnormalities as recurrent changes in B-ALL. The latter has been previously reported in infant KMT2A-r ALL [32]. Such abnormalities may emerge as novel subtype-defining genomic changes in expanded patient cohorts. Furthermore, using WGS a number of new genetic features were recently associated with B-ALL subtypes previously defined by WTS. These discoveries highlight the importance of the continual discovery element associated with comprehensive WGS analysis.
We are confident from the results presented here to recommend T-only analysis for sensitive detection of clinically-relevant genetic variants as a rapid diagnostic test for implementation of risk-directed treatment stratification. This statement is supported by the failure of only one genetic abnormality in a single patient to be called by T-only analysis. This unusual case harboured a BCR::ABL1 fusion, formed through a complex rearrangement characteristic of templated insertion. This process leads to duplicated sequence surrounding double stranded breakpoints, as previously reported in multiple myeloma, and often involving the MYC gene [37]. The breakpoints surrounding BCR::ABL1 were visible within the alignment (bam) files. Thus, due to the clinical importance of BCR::ABL1 and ABL-class subtype detection in relation to their poor prognosis [2, 11, 38] and potential treatment with tyrosine kinase inhibitor (TKI) therapy [12, 39], we are developing bespoke automated calling approaches, as we achieved for IGH::DUX4. The ability for continual optimisation of such pipelines, in response to newly identified or difficult-to-detect genetic abnormalities, emphasises the potential for WGS to keep pace in routine diagnostic testing.
IGH::DUX4 accounts for ~10% of B-other-ALL and is regarded as a genetic marker of good-risk [7, 40]. However, studies have been limited by small cohort sizes and/or heterogeneous therapies. We developed a novel automated bioinformatic pipeline to reliably identify 60 patients with DUX4-r within UKALL2003, representing the largest DUX4-r cohort within an individual clinical trial to date. Previous studies have shown that IGH::DUX4 patients have a high incidence of ERG deletions, proposed as a surrogate marker to overcome difficulties in detection of DUX4-r [10, 23, 34, 41]. As around one third of DUX4-r cases do not have detectable ERG deletions, targeted identification of DUX4-r is a priority for accurate diagnosis. Furthermore, ERG abnormalities were detected in 68% of DUX4-r cases by WGS compared to 35% of the same cohort by MLPA. In addition, we have demonstrated the improved sensitivity of WGS to detect other patterns of secondary genetic deletions that predict treatment response (IKZF1-plus, UKALL-CNA-classifier), demonstrating the versatility of WGS in comprehensive detection of all levels of risk-stratifying genetic abnormalities [33, 36].
In an era in which genomics is driving enormous scientific progress and demonstrating the potential for precision medicine, this study endorses the clinical advantage of introducing WGS as a first line diagnostic test in childhood B-ALL. While accurately detecting the range of clinically-relevant cytogenetic abnormalities, it identified an expanded list of genetic abnormalities, which may define novel subtypes or co-operating genetic abnormalities in larger collaborative studies. Although the cost and infrastructural requirements of WGS has been limiting for many countries, the decreasing prices and rapidly expanding list of genetic tests required for accurate diagnosis are making WGS a viable option for some healthcare providers. Pipelines include prioritisation of genetic abnormalities that require an immediate clinical response, while providing wider access to sequencing, demographic and treatment data to allow new clinical associations to emerge. This study validates the importance of this new diagnostic service to detect clinically actionable genetic abnormalities and build a unique and invaluable resource for developing genetic-based risk stratification algorithms in the future.
Data availability
Sequencing data have been deposited in the European Genome-phenome Archive (EGA) under the Accession Code EGAS00001006863. Alternatively these data will be made available upon request from Prof Christine Harrison (christine.harrison@newcastle.ac.uk).
References
Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541–52.
O’Connor D, Enshaei A, Bartram J, Hancock J, Harrison CJ, Hough R, et al. Genotype-specific minimal residual disease interpretation improves stratification in pediatric acute lymphoblastic leukemia. J Clin Oncol. 2018;36:34–43.
Schwab CJ, Murdy D, Butler E, Enshaei A, Winterman E, Cranston RE, et al. Genetic characterisation of childhood B-other-acute lymphoblastic leukaemia in UK patients by fluorescence in situ hybridisation and Multiplex Ligation-dependent Probe Amplification. Br J Haematol. 2021.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15.
Lilljebjorn H, Henningsson R, Hyrenius-Wittsten A, Olsson L, Orsmark-Pietras C, von Palffy S, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790.
Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296–307.
Jeha S, Choi J, Roberts KG, Pei D, Coustan-Smith E, Inaba H, et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov. 2021;2:326–37.
Schwab C, Cranston RE, Ryan S, Butler E, Winterman E, Hawking Z, et al. Integrative genomic analysis of patients with acute lymphoblastic leukaemia lacking a genetic biomarker reveals a distinctive landscape as well as clinically relevant subtypes: a retrospective analysis of data from the UKALL2003 clinical trial. Leukemia. 2022. https://doi.org/10.1038/s41375-022-01799-4.
Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331.
Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48:1481–9.
den Boer ML, Cario G, Moorman AV, Boer JM, de Groot-Kruseman HA, Fiocco M, et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol. 2021;8:e55–e66.
Moorman AV, Schwab C, Winterman E, Hancock J, Castleton A, Cummins M, et al. Adjuvant tyrosine kinase inhibitor therapy improves outcome for children and adolescents with acute lymphoblastic leukaemia who have an ABL-class fusion. Br J Haematol. 2020;191:844–51.
de Haas V, Ismaila N, Advani A, Arber DA, Dabney RS, Patel-Donelly D, et al. Initial diagnostic work-up of acute leukemia: ASCO clinical practice guideline endorsement of the College of American Pathologists and American Society of Hematology Guideline. J Clin Oncol. 2019;37:239–53.
Harrison CJ, Haas O, Harbott J, Biondi A, Stanulla M, Trka J, et al. Detection of prognostically relevant genetic abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: recommendations from the Biology and Diagnosis Committee of the International Berlin-Frankfurt-Munster study group. Br J Haematol. 2010;151:132–42.
Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science. 2022;376:44–53.
Boer JM, Marchante JRM, Evans WE, Horstmann MA, Escherich G, Pieters R, et al. BCR-ABL1-like cases in pediatric acute lymphoblastic leukemia: a comparison between DCOG/Erasmus MC and COG/St. Jude signatures. Haematologica 2015;100:E354–E7.
Brady SW, Roberts KG, Gu Z, Shi L, Pounds S, Pei D, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54:1376–89.
Duncavage EJ, Schroeder MC, O’Laughlin M, Wilson R, MacMillan S, Bohannon A, et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N Engl J Med. 2021;384:924–35.
Meggendorfer M, Jobanputra V, Wrzeszczynski KO, Roepman P, de Bruijn E, Cuppen E, et al. Analytical demands to use whole-genome sequencing in precision oncology. Semin Cancer Biol. 2022;84:16–22.
Berglund E, Barbany G, Orsmark-Pietras C, Fogelstrand L, Abrahamsson J, Golovleva I, et al. A Study Protocol for Validation and Implementation of Whole-Genome and -transcriptome sequencing as a comprehensive precision diagnostic test in acute leukemias. Front Med. 2022;9:842507.
Vora A, Goulden N, Wade R, Mitchell C, Hancock J, Hough R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14:199–209.
Vora A, Goulden N, Mitchell C, Hancock J, Hough R, Rowntree C, et al. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2014;15:809–18.
Schwab CJ, Murdy D, Butler E, Enshaei A, Winterman E, Cranston RE, et al. Genetic characterisation of childhood B‐other‐acute lymphoblastic leukaemia in UK patients by fluorescence in situ hybridisation and Multiplex Ligation‐dependent Probe Amplification. Br J Haematol 2021;196:753–63.
Schwab CJ, Jones LR, Morrison H, Ryan SL, Yigittop H, Schouten JP, et al. Evaluation of multiplex ligation-dependent probe amplification as a method for the detection of copy number abnormalities in B-cell precursor acute lymphoblastic leukemia. Genes, Chromosomes Cancer. 2010;49:1104–13.
Harrison CJ, Moorman AV, Barber KE, Broadfield ZJ, Cheung KL, Harris RL, et al. Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol. 2005;129:520–30.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2.
Roller E, Ivakhno S, Lee S, Royce T, Tanner S. Canvas: versatile and scalable detection of copy number variants. Bioinformatics. 2016;32:2375–7.
Barinka J, Hu Z, Wang L, Wheeler DA, Rahbarinia D, McLeod C, et al. RNAseqCNV: analysis of large-scale copy number variations from RNA-seq data. Leukemia. 2022;36:1492–8.
Li J-F, Dai Y-T, Lilljebjörn H, Shen S-H, Cui B-W, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sciences USA 2018;115:E11711-20.
Papaemmanuil E, Rapado I, Li YL, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116.
Sinclair PB, Ryan S, Bashton M, Hollern S, Hanna R, Case M, et al. SH2B3 inactivation through CN-LOH 12q is uniquely associated with B-cell precursor ALL with iAMP21 or other chromosome 21 gain. Leukemia 2019;33:1881–94.
Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47:330–7.
Moorman AV, Enshaei A, Schwab C, Wade R, Chilton L, Elliott A, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434–44.
Zaliova M, Potuckova E, Hovorkova L, Musilova A, Winkowska L, Fiser K, et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica. 2019;104:1407–16.
Schwab CJ, Chilton L, Morrison H, Jones L, Al-Shehhi H, Erhorn A, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. 2013;98:1081–8.
Stanulla M, Dagdan E, Zaliova M, Moricke A, Palmi C, Cazzaniga G, et al. IKZF1(plus) defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. 2018;36:1240.
Bergsagel PL, Kuehl WM. Promiscuous structural variants drive myeloma initiation and progression. Blood Cancer Discov. 2020;1:221–3.
Cario G, Leoni V, Conter V, Attarbasc A, Zaliova M, Sramkova L, et al. Relapses and treatment-related events contributed equally to poor prognosis in children with ABL-class fusion positive B-cell acute lymphoblastic leukemia treated according to AIEOP-BFM protocols. Haematologica. 2020;105:1887–94.
Schultz KR, Carroll A, Heerema NA, Bowman WP, Aledo A, Slayton WB, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group Study AALL0031. Leukemia. 2014;28:1467–71.
Li Z, Lee SHR, Chin WHN, Lu Y, Jiang N, Lim EH, et al. Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv. 2021;5:5226–38.
Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28:70–7.
Li Y, Schwab C, Ryan S, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508:98–102.
Hirabayashi S, Ohki K, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102:118–29.
Hormann FM, Hoogkamer AQ, Beverloo HB, Boeree A, Dingjan I, Wattel MM, et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica. 2019;104:e455–e9.
Russell LJ, Akasaka T, Majid A, Sugimoto KJ, Loraine Karran E, Nagel I, et al. t(6;14)(p22;q32): a new recurrent IGH@ translocation involving ID4 in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood. 2008;111:387–91.
Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBDO, Berti E, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: lymphoid neoplasms. Leukemia. 2022;36:1720–48.
Acknowledgements
Primary childhood leukaemia samples used in this study were provided by the Blood Cancer UK Childhood Leukaemia Cell Bank. We also thank all the members of the NCRI Childhood Cancer and Leukaemia Group (CCLG) Leukaemia Subgroup for access to material and data on clinical trial patients. This study was supported by Blood Cancer UK (grant 15036) and European Research Council (grant 249891). SLR is a Career Development Fellow supported by Cancer Research UK (CRUK) (grant C60802/A27193). CGM is supported by the American, Lebanese and Syrian Associated Charities of St. Jude Children’s Research Hospital, and NIH CA197695
Author information
Authors and Affiliations
Contributions
CJH, SLR, MTR, and DRB designed and coordinated the study; SLR, ZK, TJ and LJR prepared the patient samples for sequencing; SLR, JFP, ZK, JB, MM, RB and LJ Russell facilitated with the WGS analysis and the development of bioinformatic data processes;SLR, JFP, TJ, PP, MM, DJ.H, KR and CGM performed the WTS analysis and the development of methods to process the data; SLR, JP, ZK, TJ, JB, PP, MM, RY, KR CGM, DRB, CJH and MT. Ross interpreted the output from WGS and WTS analyses; CJS performed MLPA and FISH experiments, assisted by REC; AVM, provided UKCNA-ALL classifier information; AV provided clinical information; SLR, DRB, CJH and MTR accessed and verified the data and wrote the manuscript, with support from all authors. CJH and MTR share senior authorship.
Corresponding authors
Ethics declarations
COMPETING INTERESTS
MTR, DRB, JFP, ZK, MM, JB and TJ are employees of Illumina, a public company that develops and markets systems for genetic analysis. CGM has received consulting and advisory board fees from Illumina Inc. and Amgen, and research funding form Pfizer and AbbVie.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ryan, S.L., Peden, J.F., Kingsbury, Z. et al. Whole genome sequencing provides comprehensive genetic testing in childhood B-cell acute lymphoblastic leukaemia. Leukemia 37, 518–528 (2023). https://doi.org/10.1038/s41375-022-01806-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-022-01806-8
This article is cited by
-
Cas9-directed long-read sequencing to resolve optical genome mapping findings in leukemia diagnostics
Scientific Reports (2024)
-
Genomic technologies for detecting structural variations in hematologic malignancies
Blood Research (2024)
-
The role of next-generation sequencing in hematologic malignancies
Blood Research (2024)
-
Refining risk prediction in pediatric acute lymphoblastic leukemia through DNA methylation profiling
Clinical Epigenetics (2024)
-
Multimodal classification of molecular subtypes in pediatric acute lymphoblastic leukemia
npj Precision Oncology (2023)
