
Abstract
Acute myeloid leukemia (AML) is a heterogeneous hematological malignancy caused by mutations in genes encoding transcriptional and epigenetic regulators together with signaling genes. It is characterized by a disturbance of differentiation and abnormal proliferation of hematopoietic progenitors. We have previously shown that each AML subtype establishes its own core gene regulatory network (GRN), consisting of transcription factors binding to their target genes and imposing a specific gene expression pattern that is required for AML maintenance. In this study, we integrate gene expression, open chromatin and ChIP data with promoter-capture Hi-C data to define a refined core GRN common to all patients with CEBPA-double mutant (CEBPAN/C) AML. These mutations disrupt the structure of a major regulator of myelopoiesis. We identify the binding sites of mutated C/EBPα proteins in primary cells, we show that C/EBPα, AP-1 factors and RUNX1 colocalize and are required for AML maintenance, and we employ single cell experiments to link important network nodes to the specific differentiation trajectory from leukemic stem to blast cells. Taken together, our study provides an important resource which predicts the specific therapeutic vulnerabilities of this AML subtype in human cells.

Similar content being viewed by others


Introduction
AML-causing genetic abnormalities that disrupt transcription factors (TFs) such as RUNX1 and C/EBPα [1, 2] interfere with hematopoietic differentiation. Different mutations lead to different disease phenotypes, gene expression profiles and clinical outcomes [3]. We previously defined the gene regulatory networks (GRNs) of seven AML subtypes including t(8;21), Inv(16), FLT3-TKD, FLT3-ITD, NPM1, RUNX1, biallelic CEBPAN/C and CEBPA single mutations by collecting transcriptome, digital footprinting and chromatin conformation capture data [4]. We showed that each AML subtype is maintained by a distinct GRN which differs from that of normal myeloid progenitor cells, demonstrating that aberrant differentiation trajectories generated new cellular identities.
C/EBPα is essential for the generation of granulocyte-monocyte progenitors (GMPs) [5, 6]. The CEBPA gene is mutated in ~15% of all AMLs [7] with most mutations affecting either the N-terminal transactivation domain (TAD1, CEBPAN) or the C-terminal basic region leucine (bZIP) domain (CEBPAC) (Fig.1A) [8]. N-terminal mutations give rise to a premature stop codon, whereby internal re-initiation of translation results in the exclusive expression of the shorter C/EBPα-p30 isoform. C-terminal mutations alter the bZIP domain and disrupt C/EBPα DNA-binding and dimerization ability [9, 10]. About 50% of CEBPA mutant AMLs exhibit biallelic mutations with combined N-terminal and C-terminal lesions (CEBPAN/C) [11, 12].

A Representation of the wild-type and mutated C/EBPα transcription factor. Transactivation (TAD) and dimerization/DNA-binding domains are highlighted. B Hypersensitive sites fold difference between CEBPAN/C and PBSCs across a 200 bp window. Data are ranked by normalized tag counts of merged CEBPAN/C peaks over merged PBSCs peaks. TF binding motifs projected against hypersensitive sites are plotted alongside. C–E Motifs enriched in CEBPAN/C specific hypersensitive sites compared to PBSCs (C) and motif analysis in CEBPAN/C (D) and t(8;21) (E) specific accessible chromatin regions. Motifs differentially enriched between the two AML subtypes are depicted in pink and green.
Mouse models of CEBPA mutated AML showed that homozygous C-terminal mutations promote the expansion of pre-leukemic HSCs but impair myeloid differentiation, whereas N-terminal mutations do not impact on proliferation and preserve myeloid commitment. The combination of both mutations accelerates leukemogenesis and explains the CEBPAN/C prevalence [13] whereby the sole expression of the p30 isoform is sufficient to develop AML [14]. A recent study identified genomic regions bound by p30 in a mouse model of CEBPAN AML [15] but the binding pattern of p30 in C/EBPαN/C patients is unknown.
GRNs represent the regulatory connections between TFs and their target genes [16]. GRNs inference has traditionally relied on gene expression perturbation studies [17] but, more recently, connections between genes and TFs were determined by measuring TF binding directly [4, 18, 19]. Previously, we constructed a basic GRN of CEBPAN/C AML patients by linking distal cis-regulatory elements occupied by specific TF families to their rightful promoter using promoter-capture Hi-C data from other AML subtypes [4]. This work showed that the cellular identity of CEBPAC/N is based on distinct regulatory relationships. However, the C/EBPα binding pattern in patient cells remained unknown, the preliminary GRN was based on only three patients, considered only distal regulatory elements, and was not based on CEBPAN/C-specific Hi-C data. In this study, we defined the complete core GRN of CEBPAN/C AML by integrating multi-omics data from 10 patients. We identified CEBPAN/C AML-specific cis-regulatory elements and TF modules, highlight AP-1, RUNX1 and C/EBPα as crucial factors driving AML maintenance and show that mutant C/EBPα and AP-1 factors colocalize with RUNX1 in chromatin. Our study therefore provides important insights into the biology of the disease in humans.
Methods
Detailed methods can be found in the Supplementary information.
Cell lines and cell line culture
Kasumi-1 and HEK 293T cells were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen. KO52 were obtained from the Japanese Collection of Research Bioresources Cell Bank. Kasumi-1 cells were cultured in RPMI-1640 (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS) (Gibco), 5% Penicillin-Streptomycin (Pen-Strep) (Sigma-Aldrich), 5% L-Glutamine (L-Glu) (Sigma-Aldrich). HEK 293T cells were cultured in DMEM supplemented with 10% FCS (Gibco), 5% Pen-Strep, 5% L-Glu. KO52 were cultured in Alpha MEM (Lonza) supplemented with 10% FCS (Gibco), 5% Pen-Strep, 5% L-Glu.
Primary cell culture and isolation of hematopoietic stem/progenitor cells
C/EBPAN/C-8 cells were cultured at the concentration of 1 × 106 cell/mL in StemSpanTM Serum-Free Expansion Medium II (SFEMII) plus 10% StemSpanTM CD34+ Expansion Supplement and 175 nM UM171 (STEMCELL Technologies). All other patient cells were grown in SFEMII supplemented with 100 ng/mL thrombopoietin, 10 ng/mL FMS-like tyrosine kinase 3 ligand, 750 nM Stem Regenin 1 (SRI), 10 ng/mL Interleukin 3, 10 ng/mL human granulocyte/macrophage colony stimulating factor, 150 ng/mL Stem Cell Factor (all Peprotech) and UM729 (STEMCELL Technologies). Patient CD34+ or CD117+ progenitors were isolated with CD34 MicroBead Kit, human or CD117 MicroBead Kit, human (Miltenyi-Biotech) as described in [4].
dnC/EBP and dnFOS lentivirus plasmids
The doxycycline inducible pCW57.1-dnC/EBP plasmid was generated using Gateway Gene Cloning (Thermo Fisher Scientific). Plasmids containing the dnC/EBP and dnFOS inserts were a gift from Charles Vinson, National Cancer Institute, Bethesda, USA [20]. The pCW57.1 containing the dnFOS insert was generated by Dr Sandeep Potluri (University of Birmingham) following the same protocol [21].
RNA extraction, real-time PCR and RNA-seq library preparation
Total RNA was extracted with NucleoSpin RNA (Machery-Nagel) and was retrotranscribed using SuperScriptTM II Reverse Transcriptase kit (Thermo Fisher) and Oligo(dT) primers (Promega). Real-time PCR and RNA-Seq was performed as described in [4].
DNase-seq and ATAC-seq
DHS mapping was performed as described in [4]. ATAC libraries were generated following the Omni-ATAC protocol from [22].
Chromatin immunoprecipitation followed by sequencing (ChIP-seq)
Double protein–protein crosslinking followed by protein-DNA crosslinking with 0.25 M Di(N-succinimidyl) glutarate (DSG) (Sigma-Aldrich) for 45 min on rotation at RT was performed when immunoprecipitating chromatin with the c-FOS antibody, a single-crosslinking approach (protein-DNA crosslinking) in all other cases as described in [23].
Promoter-capture Hi-C (CHi-C)
CHi-C in primary AML cells was performed as described in [4].
DNase I-seq and ATAC-seq data analysis
Details of the bioinformatics analyses can be found in the Supplementary information.
Results
CEBPA N/C AML forms a distinct epigenetic sub-group as compared to other AML subtypes
In t(8;21) the ETO gene is fused to the gene encoding RUNX1, creating the fusion protein RUNX1/ETO [24]. The expression and open chromatin profiles of CEBPAN/C and t(8;21) AML clustered closely together [4]. We originally explained this feature with the observation that both mutations affect the same pathway, as CEBPA is mutated in CEBPAN/C AML and is repressed by RUNX1/ETO in t(8;21) AML [24, 25]. To substantiate that CEBPAN/C represents a specific epigenetic subclass of AML, we repeated these analyses with additional samples to include a total of 10 CEBPAN/C patients, and studied 7 t(8;21) patients. CD34+ leukemic blasts were purified for RNA-seq and ATAC-or DNase-seq experiments (Supplementary Table S1 and Supplementary Fig. S1A, B). Unsupervised clustering analysis of RNA-seq (Supplementary Fig. S1A) and ATAC/DNase-seq data (Supplementary Fig. S1B) showed that the data from all CEBPAN/C patients clustered as a unique group. To define the cis-regulatory elements distinguishing CEBPAN/C from t(8;21) AML, we compared the open chromatin profiles (DNAse-Seq and ATAC-Seq data) of t(8;21) and CEBPAN/C and also compared both AML subtypes to normal CD34+ peripheral blood stem cells (PBSCs) (Fig.1B and Supplementary Fig. S1C). Individual open chromatin patterns within each subtype were highly similar (Supplementary Fig. S1C, DNase I/ATAC-Seq panel) and thus data could be merged. When compared to each other and to PBSCs, both subtypes displayed numerous specific peaks (Supplementary Fig. S1C). Motif enrichment analysis of specific open chromatin regions revealed that only RUNX and ETS motif enrichment was shared (Fig. 1D, E). Examples of genes and accessible chromatin sites being differentially regulated between the two AML subtypes are shown in Supplementary Fig. S1D, E. Two findings were noteworthy: (i) the occurrence of a CEBP:AP-1 composite motif (Fig.1B–D), which has been shown to bind JUN-CEBP heterodimers [26], and (ii) the enrichment of motifs for POU4F1 (Fig. 1C), a transcription factor that is aberrantly expressed in both CEBPAN/C and t(8;21) AMLs [4], in the CEBPAN/C-specific peaks.
We next assessed the expression of all CEBP genes in both healthy PBSCs, CEBPAN/C and t(8;21) leukemic blast cells (Fig 2A, left panel, Supplementary Fig. S2A). All were expressed in both subtypes of AML and could potentially compensate for aberrant C/EBPα function. We then compared merged transcriptomic data from CEBPAN/C AML blasts with healthy PBSCs. In CEBPAN/C AML blasts, 1354 genes were downregulated and 357 upregulated (Fig. 2B). Patient-specific expression patterns (Supplementary Fig. S1F) and the pattern of highly up- and downregulated genes (Fig. 2C, D) were consistent amongst all 10 patients. The analysis of pathways aberrantly regulated in CEBPAN/C AML as compared to normal blast cells revealed “ECM-receptor interaction” and “Adherent junctions” as downregulated pathways, in accordance with previous observation that deregulation of stem cell-niche interactions is a hallmark of AML [27] (Supplementary Fig. S2B). Upregulated pathways included “Ribosome” (Supplementary Fig. S2C) indicating increased protein synthesis in highly proliferating leukemic blasts. Note that C/EBPα recruits PolI to stimulate rRNA synthesis in myeloid progenitors [28, 29].

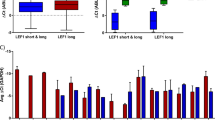
A mRNA expression levels as measured by RNA-seq of CEBPA and RUNX1 in the indicated blast types. RUNX1 expression: adjusted P value 0.0095, Limma–Voom with Benjamini–Hochberg correction for multiple testing. B Volcano plot of genes deregulated in CEBPAN/C AMLs compared to PBSCs. Each dot represents a gene, the log2 fold-change indicates the mean expression level for each gene across all CEBPAN/C samples. Blue dots represent significantly downregulated genes (log2 fold-change <–1, Benjamini–Hochberg adjusted P value <0.05); red dots represent significantly upregulated genes (log2 fold-change >1, Benjamini–Hochberg adjusted P value <0.05). C, D Hierarchical clustering of expression values for the 50 most upregulated (C) and downregulated (D) genes in CEBPAN/C AMLs compared to PBSCs.
RUNX1 was significantly downregulated in all CEBPAN/C patients except for a sample harboring a heterozygous RUNX1 292delC (Fig. 2A, right panel) carrying a premature stop codon. In contrast, the mRNA level of CEBPA was increased (Fig. 2A, left panel). CEBPAN/C showed more downregulated genes as compared to t(8;21) (Supplementary Figs. S2D–F and S3A, B) but the comparison to PBSCs showed that most downregulated genes were shared between the two subtypes (Supplementary Fig. S2G) in contrast to upregulated genes (Supplementary Fig. S2H). This result suggests that the similarities between the two subtypes may be caused by the impediment of C/EBPα function.
Mutant C/EBPα proteins colocalize with AP-1 and RUNX1 factors in human CEBPA N/C cells
CEBPAN/C AMLs express only one DNA-binding form of C/EBPα (p30) (Fig. 1A). To identify genome-wide p30 C/EBPα binding sites, we performed ChIP-seq assays in purified blast cells of four CEBPAN/C patients. Data were ranked alongside the fold-change between CEBPAN/C ATAC-Seq peaks and those of healthy PBSCs. We found that the mutant C/EBPα binding patterns were comparable in all samples and were skewed toward CEBPAN/C patient-specific accessible chromatin sites (Fig. 3A, ChIP panels). RUNX1 and C/EBPα cooperate to regulate myeloid-specific genes [30, 31]. We therefore measured RUNX1 binding and found that binding patterns in all four patients closely followed those of C/EBPα (Fig. 3A, ChIP panels). Motif alignment analysis (Fig. 3A, Motif panels) showed that C/EBPα ChIP peaks were enriched for C/EBP motifs and again showed an enrichment for C/EBP:AP-1 composite motifs only in CEBPAN/C patient samples. Mutant C/EBPα binding was associated with active and inactive genes (Fig. 3A, outermost panel).

A Density plot of C/EBPα (red), RUNX1 (green) and FOS (orange) ChIP-Seq. peaks in CEBPAN/C samples and the KO52 cell line across a 2 kb window. ChIP data, together with TF binding motifs (blue plots) and gene expression data (rightmost plot), are plotted alongside hypersensitive sites ranked by normalized tag counts of merged CEBPAN/C peaks over merged PBSCs peaks. B Venn diagram showing the number of hypersensitive sites shared between the KO52 cell line and CEBPAN/C patient samples. C, D Venn diagrams showing the number of C/EBPα (C) and RUNX1 (D) ChIP peaks shared between KO52 cell line and CEBPAN/C patient samples. E Venn diagram showing the overlap of C/EBPα, RUNX1 and c-FOS ChIP peaks in KO52 cell line.
To be able to perform biochemical experiments, we employed the CEBPAC/N AML cell line model KO52 [15, 32]. A total of 77.8% of all KO52 peaks overlapped with those from patients, demonstrating that the subtype identity of these cells is largely preserved (Fig. 3B). ChIP experiments for C/EBPα, RUNX1 and the AP-1 family member FOS showed that the pattern of binding resembled that of patients (Fig. 3A, right ChIP panel, Supplementary Fig. S3C–E for motif analyses). RUNX1 and C/EBPα ChIP-Seq peaks of patients were mostly contained within the KO52 peak population (Fig. 3C, D) with around 40% of FOS (AP-1) binding sites colocalizing with C/EBPα (Fig. 3E). The majority of RUNX1 peaks overlapped with either C/EBPα (47%) or AP-1 (60%) (Fig. 3E). RUNX1 itself was bound by all three factors sharing several binding sites (Supplementary Fig. S3F). Bootstrapping analysis, which measures the frequency of TF binding motifs colocalized within 50 bp, confirmed this result (Supplementary Fig. S3G). A detailed analysis of motif spacing revealed a specific arrangement of binding sites, with motifs for AP-1 and C/EBP as well as AP-1 and RUNX1, but not C/EBP and RUNX1 being periodically arranged with a 20 bp distance (Supplementary Fig. S3H). These arrangements were specific for ATAC sites harboring ChIP peaks (Supplementary Fig. S3I, J). In the C/EBPα/AP-1 ChIP sites we found several colocalized binding motifs, most of which turned out to be C/EBP:AP-1 sites, again suggesting factor cooperation. Integration of differentially expressed genes with ChIP data showed that in between 30 and 50% of genes upregulated in CEBPAN/C AML compared to normal cells were bound by each of the three factors (Supplementary Fig. S4A, B) but this was true only for an average of 25% of downregulated genes (Supplementary Fig. S4C, D), suggesting that this cooperation is important for the establishment of AML-specific gene activity.
The identification of a core gene regulatory network for CEBPAN/C AML
The definition of the GRN on which CEBPAN/C AMLs rely requires the assignment of regulatory sequences to their target promoters. We therefore defined the whole set of distal element-promoter interactions for one of our CEBPAN/C patient samples (CEBPAN/C-9, Supplementary Table S1) using promoter-capture Hi-C (CHi-C). The interactome for both normal CD34+ cells and t(8;21) patients was previously characterized [4, 33]. As expected, most interactions occurred within the same chromosome (Supplementary Fig. S4E) and the organization into TADs in PBSCs, CEBPAN/C and t(8;21) AMLs did not differ (Fig. 4A). However, when zooming in on individual genes, differences became apparent, as shown with the SPI1 locus (Supplementary Fig. S4F). More than 100,000 interactions between distal open chromatin regions and promoters were identified in each sample, and ~88% of CEBPAN/C interactions were also present in the t(8;21) dataset (Supplementary Fig. S5A). A similar overlap was also seen when compared to healthy CD34+ cells. To gain further insight into the biology of CEBPAN/C AML we identified the cis-regulatory elements underlying CEBPAN/C specific interactions by assigning them to open chromatin regions. 3692 DHSs were within this population (Fig. 4B). We assigned those elements to their rightful genes using CHi-C (Supplementary Dataset 1) and compared gene expression to PBSCs (Supplementary Table S2 and Supplementary Dataset 2). CEBPAN/C specific DHSs were mostly associated with downregulated genes important for myelopoiesis, including MAF and RUNX1. 72.5% of upregulated and 61.9% of downregulated genes were C/EBPα targets, demonstrating a major role of the oncogene in shaping the AML subtype-specific chromatin landscape.

A Contact matrix across chromosome 1 for healthy CD34+ blasts (top) and t(8;21) (middle) and CEBPAN/C (bottom) leukemic blasts at 10 Mb resolution. A UCSC genome browser track is shown below each matrix to highlight the DNAse I hypersensitive site pattern. B Overlap of DNase I-HS sites with interactions for t(8;21) and CEBPAN/C AML as compared to normal CD34+ cells.
To construct the CEBPAN/C-specific GRN, we first defined the potential cis-regulatory elements deregulated in leukemic blasts as compared to PBSCs (Fig. 5A) [4]. We then performed a motif search within these DHSs to infer candidate TFs that bind these sites by using a list of non-redundant motifs representing 80 TF families expressed in myeloid cells [4]. Using CHi-C data, we linked each binding motifs to the promoters of the genes that they regulate (Fig. 5B). Each network node represents a TF colored according to its expression in CEBPAN/C AMLs. TF families binding to the same motif form a composite node (encircled), arrows going from a node to a specific gene indicate the presence of a binding motif in the locus encoding that gene. The analysis highlights nodes and edges which are specific for CEBPAN/C AMLs and how different TFs and their genes are “wired”. The C/EBP and AP-1 families represent major nodes, with 341 and 353 connections to putative targets, respectively (Supplementary Dataset 3), suggesting a high regulatory relevance. ETS, RUNX and E-box factors, which are part of the global hematopoietic signature [34], are highly connected, with the RUNX family being linked to about 500 genes (Supplementary Dataset 3).

A Network construction strategy overview based on the aggregate of data from 10 patients. B TF targets determined by known motifs as described in [4]. Arrows (Edges) pointing outward from entire node highlight motifs present in individual genes whereby the hypersensitive site was annotated to the gene using the CHi-C data where possible, or otherwise to the nearest gene. The expression level of the individual genes (CPM) in CEBPAN/C samples is depicted in blue (low) or red (high) color. Edges are colored accordingly with the number of motifs. Node edge color indicates the gene expression level in CEBPAN/C samples compared to PBSCs.
Our GRN construction strategy relies on the identification of TF binding motifs. For validation, we compared C/EBP, RUNX and AP-1 targets identified in the CEBPAN/C-specific GRNs with C/EBPα, RUNX1 and c-FOS ChIP data. Almost 50% of AP-1 family targets identified in our GRNs were bound by c-FOS in KO52 cells (Supplementary Fig. S5B). The overlap was over 60% for the RUNX1 (Supplementary Fig. S5C) and over 80% for the C/EBP targets (Supplementary Fig. S5D), demonstrating the reliability of our strategy. The comparison of AML subtype-specific pathways from the three modules showed little overlap, with the most deregulated pathways in the AP-1 module being associated with signaling and cell cycle as expected (Supplementary Fig. S5B–D and Supplementary Dataset 4).
Analysis of CEBPA C/N leukemic stem cells (LSCs) and blast populations at the single cell level
LSCs are a rare subpopulation among leukemic blasts that survives chemotherapy [35]. To identify LSC-specific vulnerabilities, we characterized LSCs (CD34+ CD90– CD38–) and leukemic blasts (CD34+ CD90– CD38+) purified from a CEBPAN/C AML sample expressing both C/EBPα isoforms (Supplementary Fig. S6A–C). ATAC-seq analysis showed 3263 open chromatin sites enriched in LSCs which were associated with specific gene expression program, and 5761 enriched in blasts, (Fig. 6A, Supplementary Fig. 6D and Supplementary Dataset 5). The analysis of the gene expression profile showed an LSC score [36] of –0.22 in LSC populations compared to –0.45 in blasts, confirming the nature these cells as LSCs (Supplementary Fig. S6E). Motif analysis showed enrichment of C/EBP motifs in blast-specific sites while GATA motifs were more represented in LSC-specific sites, in accordance with their grade of differentiation (Fig. 6A). The composite C/EBP:AP-1 motif was specifically enriched in the blast-specific ATAC-Seq sites (Fig. 6A).

A ATAC-seq peak profile comparison of LSC and blast cells purified from CEBPAN/C-8 patient sample across a 2 kb window. Data are ranked by normalized tag counts of LSC peaks over blasts. TF binding motifs projected against hypersensitive sites and fold differences in gene expression associated with open chromatin regions are plotted alongside. B Analysis of scRNA-Seq data. Uniform Manifold Approximation and Projection for Dimensional Reduction (UMAP) map displaying LSC and blast clusters as identified with GSEA analysis. Each dot in the map represents a cell and is colored accordingly to cluster assignment. C Monocle pseudotime trajectory of LSC and blast cells projected on the UMAP map of scRNA clusters.
Bulk ATAC-seq analysis highlights differences in the LSC and blast motif signatures but does not take into account that each of these populations represents a continuum of differentiation stages. We thus performed single cell (sc)RNA-seq on LSCs and blasts from one CEBPAN/C patient sample. Clustering analysis revealed 8 cell clusters (Fig. 6B). We then compared the sets of marker genes from each cluster to an LSC/Blast gene expression signature derived from our bulk RNA-Seq data using GSEA, which identified three clusters as LSC, with the remaining five identified as blasts (Supplementary Fig. S6F, G). The analysis of genes expressed in different phases of the cell cycle showed that LSCs were mainly in G0/G1 phase, consistent with their quiescent state. The cell cycle status of blast cells was more variable, with the Blast-5 cluster showing the highest proliferation rate (Supplementary Fig. S6H, I). In a pseudotime analysis, the LSC-1 cluster was localized at the apex of the differentiation trajectory (Fig. 6C) with the strong enrichment of a LSC gene expression signature (Supplementary Fig. S6G). The Blast-1 population gives rise to Blast-2, which subsequently branches into other three blast populations (Fig. 6C and Supplementary Fig. S6I).
We then projected the expression of genes encoding important hematopoietic TFs that form nodes in the CEBPAN/C AML GRN on the cell clusters (Fig. 7 and Supplementary Fig. S7). As expected, CEBPA expression was increased in all blast clusters compared to LSCs. SOX4 was expressed at higher levels in the LSC compartment (Fig. 7, for statistical significance see Supplementary Fig. S8A). In this context, note that SOX4 is repressed by wild-type C/EBPα and is required for self-renewal in CEBPAN/C LSCs [37]. The aberrantly expressed TF POU4F1 is expressed only within the LSC-1 cluster (Supplementary Fig. S7). The AP-1 family member FOS is expressed across all clusters, while its partner JUN is preferentially expressed in LSCs (Supplementary Fig. S7).

Expression of indicated genes projected on the UMAP map (upper left panel). Color intensity represent expression data log2 normalized unique molecular identifier (UMI) counts.
The growth of CEBPA N/C AML is dependent on RUNX1 as well as AP-1, and C/EBP TF families
C/EBP, RUNX and AP-1 families constitute prominent nodes of the CEBPAN/C-specific GRN. To investigate to which extent these TFs sustain the aberrant leukemic program, we generated lentiviral constructs expressing doxycycline(Dox)-inducible dominant negative C/EBP (dnC/EBP) [20] and FOS (dnFOS) [38] peptides, which heterodimerize with multiple members of the C/EBP and AP-1 families, respectively, and abolish their DNA binding, or a GFP-expressing empty vector control [21]. We could not transduce dnC/EBP and dnFOS vectors into CEBPAN/C patient blasts and therefore used KO52 and the t(8;21) cell line Kasumi-1 (Supplementary Figs. S8B and S9A–D).
After Dox treatment and FACS sorting for GFP positive cells, we performed ATAC-seq and RNA-seq on transduced cells. After induction of dnC/EBP expression for 72 h in KO52 cells, ATAC analyses identified 420 lost and 702 gained open chromatin regions (Fig. 8B). Gained ATAC sites were enriched in AP-1, RUNX1, ETS and GATA motifs, whereas lost sites were enriched in PU.1/ETS, RUNX and C/EBP motifs (Fig. 8B and Supplementary Fig. S8C), suggesting that C/EBP binding was lost. However, we observed minimal changes in gene expression (Supplementary Fig. S8D) with 31.1% of upregulated and 47.4% of downregulated genes being C/EBPα targets (Supplementary Fig. S8E). ATAC-seq experiments in Kasumi-1 cells after inducing dnC/EBP revealed that 805 open chromatin regions enriched in C/EBP motifs were lost (Supplementary Fig. S9A–C), demonstrating a higher sensitivity of this AML subtype against C/EBP inhibition.

A Volcano plot of hypersensitive sites deregulated in KO52 cells expressing dnC/EBP compared to control. Blue and red dots represent significantly downregulated (log2 fold-change <–1, Benjamini–Hochberg adjusted P value <0.05) and significantly upregulated (log2 fold-change >1, Benjamini–Hochberg adjusted P value <0.05) hypersensitive sites. Right panel: motif enriched in dnC/EBP-specific and control-specific hypersensitive sites. B Density plots showing the ATAC-seq profile of KO52 cells expressing dnC/EBP or a control vector across a 2 kb window (2 replicates). Data are ranked by normalized tag counts of dnC/EBP expressing cells over control. TF binding motifs projected against hypersensitive sites are plotted alongside. C Cumulative growth time course of KO52 cells expressing either a control vector, dnC/EBP or dnFOS (N = 3). D Cumulative growth time course of KO52 cells expressing an inducible shRNA targeting RUNX1 with or without doxycycline (N = 3). In C and D, error bars represent the sample standard deviation. P values were calculated using a two-tailed t-test.
The induction of dnFOS in KO52 cells resulted in a gain of 191 open chromatin regions, which were enriched in PU.1 and ETS motifs, and loss of 857 sites with a significant enrichment of RUNX, GATA and AP-1 motifs (Supplementary Fig. S10A–C), suggesting that closing of chromatin was due to loss of AP-1 binding. Gene expression analysis of dnFOS transduced cells showed 288 differentially regulated genes, with most genes (180) being downregulated (Supplementary Fig. S10D). KEGG pathway analysis showed that various downregulated genes encode for cell signaling components, such as SRC and multiple chemokines (Supplementary Fig. S10E), together with CEBPE which is bound by FOS, C/EBPα and RUNX1 (Supplementary Fig. S10F).
To test whether C/EBPs and AP-1 family members were required for leukemic cell growth, we performed proliferation assays with KO52 and Kasumi-1 cells expressing dominant negative constructs. Both dnC/EBP and dnFOS expression decreased cell growth in our CEBPAN/C and t(8;21) cell line models ((Fig. 8C and Supplementary Fig. S9D) and [4]). Using a DOX-inducible shRNA (Supplementary Fig. S11B), we also confirmed that JUND is required for efficient proliferation of KO52 cells (Supplementary Figs. S10G and S11A). The presence of dnC/EBP and dnFOS constructs in KO52 cells led to an increased loss of GFP+ cells as compared to the induced empty vector construct (Supplementary Fig. S11B), suggesting that the expression of the peptides is toxic.
RUNX1 has been shown to be essential for the survival of t(8;21) AML [39]. We therefore depleted RUNX1 directly using a DOX-inducible shRNA (Fig. 8D and Supplementary Fig. 11C). We also employed a small molecule inhibitor of CBFβ (AI-14-91), which disrupts its interaction with the RUNT domain of RUNX proteins and has been shown to affect the survival of AML but not normal cells [23, 40] (Supplementary Fig. S10H). These experiments demonstrate that the RUNX1 inhibitor, but not the control compound AI-4-88, and RUNX1 depletion strongly reduce KO52 growth, with an increase in apoptosis (Supplementary Fig. S11D). FACS analyses demonstrated a modest change in the differentiation state of the cells, with JUND depletion increasing and RUNX1 depletion decreasing CD34+ cell numbers (Supplementary Fig. S11E–H), suggesting that the balance of RUNX1, AP-1 and mutant C/EBP impacts on the differentiation state of the cells. Taken together, these data show that our GRN analysis is predictive, demonstrating that the highly connected network nodes RUNX1, AP-1 and C/EBP are essential for regulating the growth of CEBPAN/C and t(8;21) cells.
Discussion
AP-1 and C/EBP shape the regulatory phenotype of CEBPAN/C AMLs
C/EBPα is required for myeloid differentiation and for maintaining the self-renew ability and the quiescent state of adult HSCs [41, 42]. Therefore, it was important to examine how and where mutant proteins interact with the genome and regulate gene expression. Our data suggest that mutant C/EBPα proteins cooperate with other TFs, as indicated by the presence of the composite C/EPB:AP-1 motif in CEBPAN/C-specific DHSs and the colocalization of C/EBPα, RUNX1 and AP-1, in contrast to the enrichment of the AP-1 canonical motif in t(8;21) and healthy blasts-specific sites. We showed previously that AP-1 activity is required for leukemic maintenance in FLT3-ITD and t(8;21) AMLs [4, 21, 43]. Here we show that this is also true for CEBPAN/C. C/EBP and AP-1 TFs form heterodimers [44,45,46], and C/EBPα:AP-1 interaction is required to drive monocytic lineage commitment [26]. Our data indeed suggest such collaboration. C/EBP and the C/EBP:AP-1 motif signatures were mainly confined to blast cells, suggesting that cooperation may be involved in maintaining this specific differentiation state. Our single cell experiments confirmed these differences. Except for JUN, AP-1 family members were uniformly expressed throughout all LSC and blast subpopulations. JUN was downregulated in the blast compartment, following an expression pattern inversely correlated with CEBPA. During granulocyte differentiation C/EBPα heterodimerizes with JUN, impeding the formation of JUN:FOS complexes and preventing JUN from autoregulate its own promoter. Therefore, JUN downregulation promotes granulocytic differentiation [47].
The CEBPAN/C-specific core GRN Highlights the AML-specific connections between TF-coding genes that are distinct from those found in PBSCs. The analysis of the TF modules (Supplementary Dataset 3), representing the downstream targets of TFs forming nodes, shows that these differences are responsible for the alterations in gene expression in AML.
Another important result described here is the finding that despite one essential member being mutated, the C/EBP family represents a highly connected node of the CEBPAN/C specific GRN and thus contributes to shaping the regulatory landscape. Note that a better clinical outcome of CEBPA mutant AML is associated with cells containing C/EBPα bZIP domain mutation, irrespective of whether it is co-expressed with a wt protein or a C-terminal mutant as in CEBPAN/C AML [11]. The bZIP mutant cannot bind/dimerize but could still be part of the complex and thus could interfere with multiple C/EBP proteins. Whether both mutant proteins are associated with chromatin is unclear since our antibody cannot distinguish between them. However, our colocalization studies point to an association of mutant C/EBPα proteins with other TFs which could allow some myeloid differentiation, thus improving prognosis. This finding is entirely consistent with the fact that the CEBPAN/C GRN (i) contains C/EBP proteins as a major node, (ii) that a large number of genes are bound by the mutant protein, including C/EBPAN/C-AML-specific cis-elements, (iii) that the latter are associated with downregulated myeloid differentiation genes and (iv) that eliminating this node using dnC/EBP affects the survival of the cells.
Within the CEBPAN/C GRN, the RUNX TF family is highly connected. Targets of RUNX1 in the AML-specific GRN include, for example, the apoptosis regulator BCL2 which is upregulated in CEBPAN/C AML blasts, and which could be inhibited by Venetoclax [48]. Similar to other AML subtypes [4], the AP-1 family forms an important node in the CEBPAN/C AML GRN, indicating that MAPK signaling plays an essential role in regulating growth. For FLT3-ITD and t(8;21) AML this idea has been confirmed in vitro and in vivo [4, 21].
Inhibition of the RUNX TF family and the depletion of RUNX1 shows that it is important for CEBPAN/C AML growth. Expression of dnFOS had a strong effect on the chromatin landscape of KO52 cell, mainly causing loss of open chromatin sites. Interestingly, C/EBP:AP-1 composite motifs were enriched in hypersensitive sites lost upon dnC/EBP expression in KO52, but not in Kasumi-1 cells. This result suggests that the cooperation between these TFs is a specific feature of CEBPAN/C AMLs. Our gene expression analysis showed that loss of AP-1 binding in KO52 was accompanied by downregulation of signaling pathways, suggesting that these factors promote the survival of CEBPAN/C cells. Finally, we found that the expression of dnC/EBP or dnFOS induced growth arrest in both KO52 (this study) and Kasumi-1 cells (this study and [23]).
In summary, the CEBPAN/C AML-specific core GRN highlights subtype-specific connections between TFs and target genes that code for “druggable” gene products. It will be an important resource for researchers developing combination therapies for this particular AML subtype.
Data availability
Data can be accessed at NCBI GEO (https://www.ncbi.nlm.nih.gov/geo/) under GSE211095.
References
Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–95.
Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–98.
Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–28.
Assi SA, Imperato MR, Coleman DJL, Pickin A, Potluri S, Ptasinska A, et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat Genet. 2019;51:151–62.
Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–14.
Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–74.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–70.
Wilhelmson AS, Porse BT. CCAAT enhancer binding protein alpha (CEBPA) biallelic acute myeloid leukaemia: cooperating lesions, molecular mechanisms and clinical relevance. Br J Haematol. 2020;190:495–507.
Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394–400.
Mueller BU, Pabst T. C/EBPalpha and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol. 2006;13:7–14.
Taube F, Georgi JA, Kramer M, Stasik S, Middeke JM, Rollig C, et al. CEBPA mutations in 4708 patients with acute myeloid leukemia: differential impact of bZIP and TAD mutations on outcome. Blood. 2022;139:87–103.
Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C, et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia. 2014;28:794–803.
Bereshchenko O, Mancini E, Moore S, Bilbao D, Mansson R, Luc S, et al. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell. 2009;16:390–400.
Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13:299–310.
Heyes E, Schmidt L, Manhart G, Eder T, Proietti L, Grebien F. Identification of gene targets of mutant C/EBPalpha reveals a critical role for MSI2 in CEBPA-mutated AML. Leukemia. 2021;35:2526–38.
Peter IS, Davidson EH. A gene regulatory network controlling the embryonic specification of endoderm. Nature. 2011;474:635–9.
Kucinski I, Wilson NK, Hannah R, Kinston SJ, Cauchy P, Lenaerts A, et al. Interactions between lineage-associated transcription factors govern haematopoietic progenitor states. EMBO J. 2020;39:e104983.
Collombet S, van Oevelen C, Sardina Ortega JL, Abou-Jaoude W, Di Stefano B, Thomas-Chollier M, et al. Logical modeling of lymphoid and myeloid cell specification and transdifferentiation. Proc Natl Acad Sci USA. 2017;114:5792–9.
Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90.
Olive M, Williams SC, Dezan C, Johnson PF, Vinson C. Design of a C/EBP-specific, dominant-negative bZIP protein with both inhibitory and gain-of-function properties. J Biol Chem. 1996;271:2040–7.
Martinez-Soria N, McKenzie L, Draper J, Ptasinska A, Issa H, Potluri S, et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell. 2018;34:626–42.e8
Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods. 2017;14:959–62.
Potluri S, Assi SA, Chin PS, Coleman DJL, Pickin A, Moriya S, et al. Isoform-specific and signaling-dependent propagation of acute myeloid leukemia by Wilms tumor 1. Cell Rep. 2021;35:109010.
Ptasinska A, Assi SA, Mannari D, James SR, Williamson D, Dunne J, et al. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia. 2012;26:1829–41.
Westendorf JJ, Yamamoto CM, Lenny N, Downing JR, Selsted ME, Hiebert SW. The t(8;21) fusion product, AML-1-ETO, associates with C/EBP-alpha, inhibits C/EBP-alpha-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol. 1998;18:322–33.
Cai DH, Wang D, Keefer J, Yeamans C, Hensley K, Friedman AD. C/EBP alpha:AP-1 leucine zipper heterodimers bind novel DNA elements, activate the PU.1 promoter and direct monocyte lineage commitment more potently than C/EBP alpha homodimers or AP-1. Oncogene. 2008;27:2772–9.
Majeti R, Becker MW, Tian Q, Lee TL, Yan X, Liu R, et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci USA. 2009;106:3396–401.
Muller C, Bremer A, Schreiber S, Eichwald S, Calkhoven CF. Nucleolar retention of a translational C/EBPalpha isoform stimulates rDNA transcription and cell size. EMBO J. 2010;29:897–909.
Antony C, George SS, Blum J, Somers P, Wu-corts D, Pimkin M. et al. CEBPA directly binds ribosomal DNA and promotes ribosomal RNA transcription in myeloid progenitors. Blood. 2021;138(Supplement 1):3269 .
Petrovick MS, Hiebert SW, Friedman AD, Hetherington CJ, Tenen DG, Zhang DE. Multiple functional domains of AML1: PU.1 and C/EBPalpha synergize with different regions of AML1. Mol Cell Biol. 1998;18:3915–25.
Fujimoto T, Anderson K, Jacobsen SE, Nishikawa SI, Nerlov C. Cdk6 blocks myeloid differentiation by interfering with Runx1 DNA binding and Runx1-C/EBPalpha interaction. EMBO J. 2007;26:2361–70.
Broad D. Depmap achilles 20q1 public. Cambridge, MA: Broad Institute; 2020.
Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. 2015;47:598–606.
Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–44.
Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–21.
Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–7.
Zhang H, Alberich-Jorda M, Amabile G, Yang H, Staber PB, Di Ruscio A, et al. Sox4 is a key oncogenic target in C/EBPalpha mutant acute myeloid leukemia. Cancer Cell. 2013;24:575–88.
Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem. 1997;272:18586–94.
Loke J, Assi SA, Imperato MR, Ptasinska A, Cauchy P, Grabovska Y, et al. RUNX1-ETO and RUNX1-EVI1 differentially reprogram the chromatin landscape in t(8;21) and t(3;21) AML. Cell Rep. 2017;19:1654–68.
Illendula A, Gilmour J, Grembecka J, Tirumala VSS, Boulton A, Kuntimaddi A, et al. Small molecule inhibitor of CBFbeta-RUNX binding for RUNX transcription factor driven cancers. EBioMedicine. 2016;8:117–31.
Hasemann MS, Lauridsen FK, Waage J, Jakobsen JS, Frank AK, Schuster MB, et al. C/EBPalpha is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet. 2014;10:e1004079.
Ye M, Zhang H, Amabile G, Yang H, Staber PB, Zhang P, et al. C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol. 2013;15:385–94.
Ptasinska A, Pickin A, Assi SA, Chin PS, Ames L, Avellino R, et al. RUNX1-ETO depletion in t(8;21) AML leads to C/EBPalpha- and AP-1-mediated alterations in enhancer-promoter interaction. Cell Rep. 2019;29:2120.
Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci USA. 1991;88:3720–4.
Hsu W, Kerppola TK, Chen PL, Curran T, Chen-Kiang S. Fos and Jun repress transcription activation by NF-IL6 through association at the basic zipper region. Mol Cell Biol. 1994;14:268–76.
Gombart AF, Grewal J, Koeffler HP. ATF4 differentially regulates transcriptional activation of myeloid-specific genes by C/EBPepsilon and C/EBPalpha. J Leukoc Biol. 2007;81:1535–47.
Rangatia J, Vangala RK, Treiber N, Zhang P, Radomska H, Tenen DG, et al. Downregulation of c-Jun expression by transcription factor C/EBPalpha is critical for granulocytic lineage commitment. Mol Cell Biol. 2002;22:8681–94.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8.
Acknowledgements
This work was funded by a studentship/development grant from Cancer Research UK, grants from the Medical Research Council (MR/S021469/1) and Blood Cancer UK (20006). We thank Genomics Birmingham for NGS sequencing and Mary Clarke for cell sorting.
Author information
Authors and Affiliations
Contributions
AA, PC, SP, LA and AP performed experiments. DC and SGK supervised experiments. PK and SAA performed data analysis. HRD provided samples. PNC supervised experiments and helped writing. CB conceived the study, supervised experiments and together with AA wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adamo, A., Chin, P., Keane, P. et al. Identification and interrogation of the gene regulatory network of CEBPA-double mutant acute myeloid leukemia. Leukemia 37, 102–112 (2023). https://doi.org/10.1038/s41375-022-01744-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-022-01744-5
This article is cited by
-
Leukemic stem cells activate lineage inappropriate signalling pathways to promote their growth
Nature Communications (2024)
-
CEBPA mutations in acute myeloid leukemia: implications in risk stratification and treatment
International Journal of Hematology (2024)
-
A C/ebpα isoform specific differentiation program in immortalized myelocytes
Leukemia (2023)
