
Abstract
Mitophagy removes dysfunctional mitochondria and is known to play an important role in the pathogenesis of several diseases; however, the role of mitophagy in acute respiratory distress syndrome (ARDS) remains poorly understood. While we have previously demonstrated that polydatin (PD) improves lipopolysaccharide (LPS)-induced ARDS, the specific mechanism remains unclear. In present study, we aimed to determine whether PD activates Parkin-dependent mitophagy to protect against LPS-induced mitochondria-dependent apoptosis and lung injury. To establish the ARDS model, C57BL/6 mice were intratracheally injected with LPS (5 mg/kg) in vivo and Beas-2B cells were exposured to 0.5 mM LPS in vitro. Our results indicate that PD facilitates Parkin translocation to mitochondria and promotes mitophagy in ARDS-challenged mice and LPS-treated Beas-2B cells. However, PD-induced mitophagy was suppressed in Parkin−/− mice and Parkin siRNA transfected cells, indicating that PD activates Parkin-dependent mitophagy. Furthermore, the protective effects of PD against LPS-induced mitochondria-dependent apoptosis and lung injury were suppressed when Parkin was depleted both in vivo and in vitro. The inhibition of mitophagy with mitophagy inhibitor mitochondrial division inhibitor-1 in vivo and silencing of autophagy-related gene 7 in vitro also blocked the protective effects mediated by PD. Our data suggest that Parkin-dependent mitophagy induced by PD provides protection against mitochondria-dependent apoptosis in ARDS.
Similar content being viewed by others

Introduction
Acute respiratory distress syndrome (ARDS) is a devastating disorder that occurs most frequently due to severe bacterial pneumonia or sepsis, and presents a critical health concern that results in high mortality rates [1, 2].
Autophagy is an intracellular self-degradation process in which double-membraned organelles, termed autophagosomes, deliver cytoplasmic materials to lysosomes [3, 4]. Mitophagy refers to the specific autophagic turnover of mitochondria, and represents an important mechanism of mitochondrial quality control [5, 6]. Moreover, mitophagy selectively removes dysfunctional or damaged mitochondria, maintains a healthy population of mitochondria, and largely plays a cytoprotective role in the context of disease pathogenesis [7, 8]; however, the specific role of mitophagy in ARDS remains poorly understood.
Parkin signalling is a key pathway involved in the regulation of mitophagy [9, 10]. In Parkin-mediated mitophagy, the depolarized mitochondria stabilize phosphatase and tensin homologue (PTEN)-induced putative kinase 1 (PINK1) on the outer mitochondrial membrane, which subsequently recruits the E3 ubiquitin ligase Parkin to initiate the autophagic degradation of damaged mitochondria [11, 12]. Although most studies on Parkin-dependent mitophagy have been related to neurodegenerative diseases (e.g., Parkinson’s disease) [8, 13], a previous study found that mitophagy acts as an important modulator in human pulmonary diseases and could represent a potential therapeutic target [14,15,16]. Therefore, we investigated the role of Parkin-dependent mitophagy as a protective mechanism against mitochondria-dependent apoptosis in LPS-induced ARDS.
Polydatin (PD) is a monocrystalline drug that can be isolated from the herb Polygonum cuspidatum, and is used for the treatment of sepsis, burns and ischaemia-reperfusion injury [17,18,19]. In the previous study, we demonstrated that PD improves lipopolysaccharide (LPS)-induced ARDS by inhibiting apoptosis in rats [20]. However, the mechanism by which PD protects against apoptosis remains unknown. In present study, we evaluated the ability of PD against LPS-induced mitochondria-dependent apoptosis in ARDS and elucidated whether its effect was related to the activation of Parkin-dependent mitophagy.
Materials and methods
Cell culture, stimulation and cell viability
BEAS 2B cells were grown at 37 °C in 5% CO2 in Dulbecco’s modified minimum essential medium (DMEM) containing low glucose, penicillin (100 U/mL), streptomycin (100 U) and 10% foetal bovine serum. The cells were stimulated in 0.5 mM LPS for 6 h to establish LPS-induced ARDS in vitro.
Cell viability was determined by the 3-[4, 5-dimethylthiazol-2-yl]−2, 5-diphenyltetrazolium bromide assay (MTT) after 6 h post LPS stimulation. Cells were incubated with 0.5 mg/ml MTT at 37 °C for 4 h. Absorbance at 570 nm was determined using an automatic microplate reader (SpectraMax M5; Molecular Devices, Sunnyvale, CA, USA).
Animal model
The experimental protocols were approved by the Animal Care and Use Committee of the Southern Medical University, Guangzhou, China. The care of animals was performed in accordance with both the National Institutes of Health and Chinese National guidelines. Adult male C57BL/6 mice weighing 22–25 g were purchased from the Experimental Animal Center at South Medical University in Guangzhou, China. The Parkin−/− mice (C57BL/6 background) were purchased from the Cyagen Biosciences Inc (Suzhou, China). All animals were allowed to acclimatize for 1 week before being used and had ad libitum access to food and water.
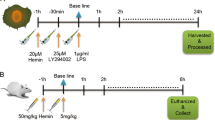
As previously described [20], ARDS was induced by an intratracheal administration of LPS. Briefly, the animals were intramuscularly anesthetized with an injection of sodium pentobarbital (30 mg/kg). The mice were placed in the supine position on a warming device, and the trachea was surgically exposed via a cervical middle line incision in the skin. LPS (5 mg/kg body weight, dissolved in 0.2 mL sterile normal saline (NS)) was slowly injected into the trachea of each mouse. Control mice were intratracheally administered 0.2 mL NS. The mice were killed 12 h post LPS administration.
siRNA transfection
siRNA targeting Parkin and Atg7 were purchased from the Santa Cruz Biotechnology. Scrambled non-targeting siRNA was used as a control. siRNA transfection was performed in accordance with the manufacturer’s protocol. Briefly, cells in the exponential phase of growth were plated in six-well tissue culture plates at a density of 1 × 105 cells per well, grown for 24 h and transfected with siRNA using Oligofectamine and OPTI-MEMI-reduced serum medium. A scrambled siRNA sequence was used as the control.
Detection of mitophagy
The pH-sensitive fluorescent protein, Keima, was used as a specific probe to quantify mitophagy. The excitation spectrum of Keima shifts from 440 to 586 nm when mitochondria are delivered to acidic lysosomes, which enables quantification of mitophagy. Images were acquired using a laser scanning confocal microscope (LSM780; Zeiss Microsystems, Jena, Germany) using the laser lines 458 nm (green, mitochondria at neutral pH) and 561 nm (red, mitochondria under acidic pH). Then the red signal areas and green areas were calculated, and the ratio (red signal area/green area) was used as an index of mitophagic activity.
Immunofluorescent assay
After treatment, the cells were fixed in a 4% paraformaldehyde solution. The cells were then blocked and incubated with the primary antibodies (anti-TOM20 and anti-Parkin) overnight at 4 °C. After rinsing with PBS, the cells were incubated with FITC or a PE-conjugated secondary fluorescent antibody for 2 h at room temperature. Images were captured using a confocal microscope (LSM780; Zeiss Microsystems, Jena, Germany).
Measurement of mitochondrial membrane potential (MMP)
The mitochondrial membrane potential (MMP) was determined using the potential-sensitive fluorescent dye JC-1. The JC-1(5 μmol/L) was loaded onto cells for 15 min at 37 °C. The results were visualized using a confocal microscope (LSM780; Zeiss Microsystems, Jena, Germany).
Western blotting
The cells and lung tissues were homogenized in ice-cold tissue lysis buffer. The homogenates were centrifuged after 30 min pyrolysis at 10,000×g for 20 min at 4 °C. For cytosolic and mitochondrial protein extraction, an isolation kit was used in accordance with the manufacturer’s instructions. The protein concentration of the supernatant was determined using the BCA method. The protein was separated on SDS-PAGE gels and transferred to polyvinylidenedifluoride (PVDF) membranes for immune blotting. The membrane was then blocked with a blocking solution (5% skimmed milk) at room temperature for 2 h, followed by an overnight incubation at 4 °C with anti-COX4I1 (1:1000 dilution), TIM23 (1:1000 dilution), PGC-1α (1:1000 dilution), mt-TFA (1:1000 dilution), Parkin (1:500 dilution), LC3 (1:500), cytochrome c (1:1000), caspase-3 (1:500 dilution), Bax (1:1000 dilution) and Bcl-2 (1:1000 dilution) monoclonal antibodies. Subsequently, the membranes were incubated with secondary antibodies (1:5000 dilution) at room temperature for 2 h. Immunoreactivity was detected with an enhanced chemiluminescence detection system (Beyotime, Haimen, China) and visualized on X-ray film (Kodak, Shanghai, China). GAPDH (1:1000 dilution) and TOM20 (1:1000 dilution) were used as controls in the cytoplasmic and mitochondrial fractions, respectively.
Measurement of cellular apoptosis
Cellular apoptosis was detected with an Annexin V-FITC apoptosis detection kit. After treatment with LPS, Beas-2B cells were washed twice with PBS and suspended in 1X binding buffer at a concentration of approximately 1 × 105 cells/mL. Then, 5 μL FITC-Annexin V and 10 μL propidium iodide was added to the cell suspension. After incubating at room temperature for 20 min in the dark, the fluorescence of the cells was determined immediately using a flow cytometer (Becton Dickinson FACScan, San Jose, CA).
Histological evaluation and scoring
Lungs were collected at 12 h after LPS injection. The right middle lobes of the lungs were fixed in 10% formalin, embedded in paraffin and sectioned to 4-μm thickness. After deparaffinization and rehydration, the sections were stained with hematoxylin and eosin. Evaluation was based on the following criteria: i) Neutrophil infiltration; ii) airway epithelial cell damage; iii) interstitial oedema; iv) hyaline membrane formation; and v) haemorrhage. Each section had five scores corresponding to the following five criteria as determined by the degree of deterioration: Normal = 0; minimal alteration = 1; mild alteration = 2; moderate alteration = 3; and severe alteration = 4. The lung injury score (LIS) for each criterion was recorded.
Immunohistochemistry
Bax and Bcl-2 expression in the lungs were visualized using immunohistochemical kits. The working dilution of antibodies against Bax and Bcl-2 was 1:200.
Measurement of the wet lung/dry lung weight (W/D) ratio
The collected wet lung was weighed and subsequently placed in an oven for 48 h at 80 °C and then weighed again when it was dry. The ratio of the wet lung to the dry lung weight was calculated.
Statistical analysis
All variables are presented as the mean ± s.d. Differences between groups were determined using a one-way ANOVA with the LSD multiple-comparison test and Student’s t-test where appropriate. Values were considered significant when P < 0.05.
Materials
PD was supplied by Neptunus Co. (Shenzhen, Guangdong, China), and MitoProbe™ JC-1 (5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide) was purchased from Molecular Probes (Invitrogen, CA). Mitochondria-targeted Keima plasmid (mt-Keima-COX8) was purchased from Public Protein/Plasmid Library (Nanjing, China). siRNA targeting Parkin and Atg7 were purchased from Santa Cruz Biotechnology. The terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) staining kit was supplied by Promega Corp. (Madison, WI). A mitochondrial/cytosolic protein extraction kit was purchased from BestBio Co. (Beijing, China). Antibodies against Parkin, COX4I1, TOM20, TIM23, PGC-1α, mt-TFA, LC3I/LC3II, Bax, Bcl-2, cytochrome c and GAPDH were obtained from Abcam (Cambridge, UK). The Caspase-3 activity assay kit was obtained from Biovision (San Francisco, USA). Immunohistochemical kits were provided by EnVision™ (Dako, Copenhagen, Denmark). The fluorescein isothiocyanate (FITC) Annexin V apoptosis kit was obtained from BD Biosciences (San Jose, CA). Human lung epithelial cell lines (Beas-2B) were obtained from Guangzhou Cellcook Biotech Co., Ltd (Guangzhou, China). All other chemicals were acquired from Sigma-Aldrich (Saint Louis, MO, USA).
Results
PD activates mitophagy in ARDS both in vivo and in vitro
To explore whether PD activates mitophagy in ARDS, mice were subjected to an intratracheal administration of LPS (5 mg/kg) and treated with PD (45 mg/kg) or vehicle (DMSO), then killed at 12 h following the administration of LPS. The level of the mitochondrial markers, COX4I1 and TIM23, were used as indicators of mitophagy activity. We found that PD treatment significantly decreased the levels of COX4I1 and TIM23 in ARDS-challenged mice, whereas the expression of the mitochondrial biogenesis marker proteins, PGC-1α and mtTFA, were not significantly downregulated with PD treatment (Fig. 1a). Moreover, PD did not significantly upregulate the level of LC3II or downregulate P62, suggesting that autophagic flux was not robustly reinforced. These results show that although PD led to a decreased accumulation of mitochondria due to mitophagy, it did not inhibit biogenesis or upregulate general autophagy.

PD activates mitophagy in ARDS both in vivo and in vitro. a Mice were subjected to an intratracheal administration of LPS (5 mg/kg) and treated with PD (45 mg/kg) or a vehicle, and killed after 12 h following LPS. Mitochondrial markers, COX4I1 and TIM23, mitochondrial biogenesis marker proteins, PGC-1α and mtTFA, autophagy-related proteins, LC3II/I and P62, were measured by western blot. b Quantification analysis of COX4I1 levels; c quantification analysis of PGC-1α levels; d quantification analysis of TIM23 levels; e quantification analysis of mt-TFA levels. f Beas-2B cells transfected with Beas-2B cells transfected with mtKeima-COX8 for 24 h were incubated with LPS (0.5 mM) and PD (50 μM) for 6 h. Laser confocal-scanning microscopy was conducted to observe mitochondria-localised Keima (green) and acidic Keima puncta (red) delivered within autolysosomes. Scale bar: 10 μm. g Quantification of acidic Keima per cell. h Quantification analysis of LC3II/I levels. i Quantification analysis of P62 levels. Data are presented as the mean ± SD (n = 6 in each group). *P < 0.05 vs. the indicated groups; #P > 0.05 vs. the indicated groups
We next investigated PD-mediated activation of mitophagy in vitro. Beas-2B cells were treated with PD (50 μM) and LPS (0.5 mM) for 6 h. To monitor mitophagy in epithelial cells, we used the pH-sensitive fluorescent protein, Keima. When Keima is present in a neutral environment (e.g., the cytoplasm or mitochondrial matrix), it is emits green fluorescence. Interestingly, when Keima is present in acidic conditions (e.g., autolysosomes), it is emit red fluorescence. As shown in Fig. 1f, PD treatment increased the ratio of red area/green area in LPS-treated cells compared with those that did not receive PD treatment, suggesting enhanced mitophagy. Taken together, these data suggest that PD activates mitophagy in ARDS.
Parkin is required for the activation of PD-induced mitophagy both in vivo and in vitro
Parkin plays a key role in the mitophagy process [10]. In Parkin-mediated mitophagy, Parkin translocation from the cytoplasm to mitochondria initiates the process [9]; however, whether Parkin is required for PD-induced mitophagy remains unclear. To determine whether Parkin is involved in the PD-induced mitophagy, we investigated the translocation of Parkin in PD-treated wild-type and Parkin−/− mice. Mice were subjected to ARDS and treated with either PD (45 mg/kg) or vehicle for 12 h. In the present study, we determined the intracellular distribution of Parkin by separating the mitochondrial and cytosolic fractions of the lungs. We found that PD treatment increased the level of mitochondrial Parkin in parallel with decreased cytosolic levels in ARDS-challenged mice compared with those without PD treatment. This finding suggested that PD increased Parkin translocation to the mitochondria in ARDS (Fig. 2a). To determine whether PD facilitates Parkin translocation to mitochondria in vitro, we co-stained Parkin and the mitochondrial marker, TOM20, in Beas-2B cells. Cells were exposed to LPS (0.5 mM) and treated with either PD (50 μM) or the vehicle for 6 h. We found that there was an elevated overlap between TOM20 and Parkin in PD-treated cells compared with those without PD treatment following LPS exposure, suggesting Parkin translocation (Fig. 2d).

PD facilitates Parkin translocation from the cytoplasm to mitochondria both in vivo and in vitro. a Mice were subjected to an intratracheal administration of LPS (5 mg/kg) and treated with either PD (45 mg/kg) or a vehicle, and killed after 12 h following LPS. The mitochondrial and the cytosolic fractions of the lung was separated, and the mitochondrial and cytosolic Parkin levels were evaluated by western blot. b Quantification analysis of cytoplasmic Parkin levels. c Quantification analysis of mitochondrial Parkin levels. d Beas-2B cells were exposed to LPS (0.5 mM) and treated with PD (50 μM) or a vehicle for 6 h. The cells were co-stained with both Parkin and the mitochondrial marker, TOM20. Scale bar: 10 μm. Data are presented as the mean ± SD (n = 6 in each group). *P < 0.05 vs. the indicated groups
To further explore the role of Parkin in PD-induced mitophagy, Parkin was downregulated both in vivo and in vitro. Wild-type or Parkin−/− mice were subjected to ARDS, and subsequently treated with either PD or a vehicle for 12 h. We found that among the mice that received PD treatment following ARDS, the level of the mitochondrial markers, COX4I1 and TIM23, were significantly increased in the Parkin−/− mice compared with wild-type mice, indicating that PD-induced mitophagy was inhibited by the knockdown of Parkin (Fig. 3a). In vitro, Beas-2B cells were transfected with Parkin siRNA (a scrambled siRNA was used as the control) for 24 h. Then the cells were exposed to LPS (0.5 mM) and treated with PD (50 μM) or a vehicle for 6 h. We found that among the LPS-exposed cells, the ratio of Keima red area/green area was decreased in the PD-treated cells with Parkin depletion compared with those without Parkin depletion, indicating a suppression of mitophagy (Fig. 3f). These data indicate that PD enhances Parkin-dependent mitophagy in ARDS.

Parkin is required for the activation of PD-induced mitophagy both in vivo and in vitro. a Wild-type or Parkin−/− mice were subjected to ARDS, and then treated with PD or a vehicle for 12 h. Mitochondrial markers, COX4I1 and TIM23, as well as mitochondrial biogenesis marker proteins, PGC-1α and mtTFA, were measured by western blot. b Quantification analysis of COX4I1 levels. c Quantification analysis of PGC-1α levels. d Quantification analysis of TIM23 levels. e Quantification analysis of mt-TFA levels. f Beas-2B cells were transfected with Parkin siRNA (a scrambled siRNA was used as a control) and mt-Keima-COX8 for 24 h. The knockdown of Parkin expression was confirmed by western blot. The cells were subsequently exposed to LPS (0.5 mM) and treated with either PD (50 μM) or a vehicle for 6 h. Laser confocal-scanning microscopy was performed to observe mitochondria-localised Keima (green) and the acidic Keima puncta (red) delivered within autolysosomes. Scale bar: 10 μm. g Quantification of the acidic Keima per cell. Data are presented as the mean ± SD (n = 6 in each group). *P < 0.05 vs. the indicated groups; #P > 0.05 vs. the indicated groups
The inhibition of mitophagy blocks PD-induced protection in mitochondria-dependent apoptosis and lung injury both in vivo and in vitro
Mitophagy largely plays a cytoprotective role in the context of disease pathogenesis [21]. However, whether the Parkin-dependent mitophagy has a protective effect in the context of ARDS has not been previously reported. To clarify the involvement of mitophagy activation on PD-mediated protection in vitro, the activity of the mitochondria-dependent apoptotic signalling pathway was assessed after inhibiting mitophagy by silencing Parkin and Atg7 (Atg7 is an essential gene for the induction of autophagy [13]. Parkin and Atg7 silencing in Beas-2B cells (a scrambled siRNA was used as control) were exposed to LPS (5 mM) and treated with either PD (50 μM) or a vehicle for 6 h. We found that PD treatment substantially inhibited the LPS-induced activation of mitochondria-dependent apoptotic signalling, as reflected by the downregulation of Bax and upregulation of Bcl-2, recovered MMP dissipation, reduction in cytochrome c release, caspase-3 activation and cellular apoptosis, and recovered cell viability (Fig. 4 and Figure s1). However, the protective effect of PD treatment against LPS-induced mitochondria-dependent apoptosis was reversed by silencing Parkin and Atg7. These data indicate that the inhibition of mitophagy blocks the PD-induced protective effects against mitochondria-dependent apoptosis.

The inhibition of mitophagy blocks PD-induced protection against mitochondria-dependent apoptosis in vitro. Parkin and Atg7 silencing of Beas-2B cells (a scrambled siRNA was used as control) were exposed to LPS (5 mM) and treated with either PD (50 μM) or a vehicle for 6 h. a The cells were then stained with JC-1 and the mitochondrial membrane potential (MMP) was observed using laser confocal-scanning microscopy. Scale bar: 20 μm. b The expression of Bax, Bcl-2 and cytoplasmic cytochrome c were detected by western blot. c Quantification of the intracellular red and green fluorescence of JC-1. d The level of caspase-3 activity was measured using the caspase-3/CPP32 fluorometric assay kit. e Cell apoptosis was evaluated with an Annexin V-FITC apoptosis detection kit. f Quantification analysis of Bax levels. g Quantification analysis of Bcl-2 levels. h Quantification analysis of cytoplasmic cytochrome c levels. Data are presented as the mean ± SD (n = 6 in each group). *P < 0.05 vs. the indicated groups
Using in vivo experiments, mdivi-1 was used to block mitophagy [22]. Mdivi-1 blocks mitochondrial fission and makes it difficult for mitochondria that have not undergone fission to be engulfed by phagophores, the precursors to autophagosomes [13]. Wild-type mice were subjected to ARDS, and then treated with a vehicle, PD (45 mg/kg), or PD simultaneously with mdivi-1 (3 mg/kg). Parkin−/− mice were subjected to ARDS and PD (45 mg/kg) treatment and sacrificed 12 h following LPS delivery. As shown in Fig. 5, PD inhibited LPS-induced mitochondria-dependent apoptosis, as reflected by the downregulation of Bax, upregulation of Bcl-2, decreased cytochrome c release, caspase-3 activation and cellular apoptosis. Moreover, there was a reduction in the extent of lung injury in these mice, as reflected by decreased histopathologic deterioration and lung oedema. However, such protection induced by PD was reversed by mdivi-1 treatment. Moreover, increased activation of mitochondria-apoptotic signalling and lung injury were detected in ARDS-challenged Parkin−/− mice, even in those that received PD treatment compared with the PD-treated wild-type mice subjected to ARDS. These results suggest that PD treatment inhibits LPS-induced mitochondria-dependent apoptosis and lung injury via the activation of Parkin-dependent mitophagy.

The inhibition of mitophagy blocks PD-induced protection against mitochondria-dependent apoptosis and lung injury in vivo. Wild-type mice were subjected to ARDS, and subsequently treated with a vehicle, PD (45 mg/kg) or PD simultaneously with mdivi-1 (3 mg/kg). Parkin−/− mice were subjected to ARDS and PD (45 mg/kg) treatment. The mice were killed 12 h following the administration of LPS. a Histological examinations were evaluated by HE-staining (×200); The pulmonary cell apoptosis in mice were measured by Tunel staining (×200); Expression of Bax and Bcl-2 were measured by immunohistochemistry (×200). b Cytoplasmic cytochrome c was detected by western blot. c Quantification analysis of cytoplasmic cytochrome c levels. d The level of caspase-3 activity was measured using a caspase-3/CPP32 fluorometric assay kit. e Graphical representation of Tunel-positive cells averaged over 10 microscopic fields per animal. f Lung oedema was detected by the wet lung/dry lung weight (W/D) ratio. g Lung injury score. Data are presented as the mean ± SD (n = 6 in each group). *P < 0.01 vs. the indicated groups
Discussion
In our previous study [20], although we demonstrated that PD improves LPS-induced ARDS, the associated mechanism remains unclear. Thus, in the present study, we demonstrated that PD provides protection against LPS-induced mitochondria-dependent apoptosis via activation of Parkin-dependent mitophagy in ARDS. Specifically, we found that PD treatment significantly decreased the levels of COX4I1 and TIM23 but did not significantly upregulate the level of LC3II. This loss of mitochondrial mass may be due to mitophagy, or alternatively, could be caused by decreased mitochondrial biogenesis. Thus, we assessed the levels of mitochondrial biogenesis marker proteins PGC-1a and mtTFA. However, none of these mitochondrial biogenesis-related proteins was significantly downregulated following PD treatment. These data suggest that PD reduces the mitochondrial mass by activating mitophagy in ARDS. Mitophagy was detected in vitro using the pH-sensitive fluorescent protein, Keima, a pH-sensitive protein that emitting green fluorescence in the cytoplasm or mitochondrial matrix, and emitting red fluorescence under acidic conditions, such as that found in autolysosomes. In LPS-exposed Beas-2B cells, we found that PD treatment was associated with an increased Keima red signal, indicating enhanced mitophagy.
Parkin-dependent signalling is a critical pathway involved in the regulation of mitophagy [10, 23]. However, it is unclear that whether the Parkin-dependent mitophagy is involved in PD-induced mitophagy. In present study, we found that PD-induced mitophagy was largely impaired in Parkin−/− mice, as reflected by the lack of reduced COX4I1 and TIM23 protein levels. Similarly, silencing Parkin with siRNA suppressed the PD-induced increase in Keima red signal area in Beas-2B cells. These results indicate that Parkin is required for the activation of PD-induced mitophagy. In the Parkin-dependent mitophagy pathway, Parkin senses a loss in mitochondrial transmembrane potential and accumulates in the damaged mitochondria [24]. Parkin, an E3 ligase, induces mitophagy by promoting mitochondrial fission to isolate the damaged mitochondrial fragments, and by ubiquitinating mitochondrial proteins to facilitate their recognition and recruitment to the autophagosomal surface [25]. Consistent with this, following PD treatment, we observed Parkin translocation both in vivo and in vitro, further supporting the activation of mitophagy by PD. We found that PD treatment increased the co-localisation of Parkin and TOM20 in vitro, which paralleled the mitochondrial Parkin levels in vivo, indicating that PD accelerates Parkin translocation to mitochondria. For the first time, these results show that Parkin-dependent mitophagy can be selectively activated by PD.
Mitophagy, often triggered by mitochondrial damage, is responsible for the elimination of dysfunctional or impaired mitochondria [26, 27]. Most studies consider the mitophagic response to mitochondrial damage to be a pro-survival mechanism [28, 29]. In our previous study, we demonstrated that PD attenuates ARDS by reducing apoptosis [20]; however, the mechanism remains unclear. In present study, we hypothesised that PD mediates its protective effects against mitochondria-dependent apoptosis by upregulating mitophagy. Moreover, such mitochondria-dependent apoptosis is mediated by mitochondrial activation, resulting in the subsequent release of cytochrome c and second mitochondrial derived activator of caspases (smac), which ultimately leads to caspase-3 activation [7, 30, 31]. To investigate the role of Parkin-dependent mitophagy on PD-induced protection in ARDS, we assessed the effects of mitochondria-dependent apoptosis while inhibiting mitophagy. Previous studies have reported that the inhibition of Atg7, a gene essential for the induction of autophagy, blocks mitophagy [13, 32]. Therefore, we inhibited Parkin-dependent mitophagy by silencing Parkin and Atg7 in vitro. We found that treatment with PD inhibited LPS-induced upregulation of Bax, downregulation of Bcl-2, MMP depolarization, cytochrome c release, caspase-3 activation, cellular apoptosis and reduction of cell viability. However, the protective effects of PD against mitochondria-dependent apoptosis were ablated by depleting Parkin and Atg7. To further confirm the role of mitophagy on PD-induced protection against lung injury, we used mdivi-1, a known inhibitor of mitochondrial fission, to inhibit mitophagy in vivo [33, 34]. Recent studies have suggested that mdivi-1 acts as an inhibitor of mitophagy by blocking autophagosome/mitophagosome formation [11, 13]. We found that mdivi-1 inhibits the protective effects of PD against LPS-induced mitochondria-dependent apoptosis, reflected by the upregulation of Bax, downregulation of Bcl-2, cytochrome c release, caspase-3 activation and cellular apoptosis. Moreover, there was reduced lung injury, as reflected by the level of histopathologic deterioration and lung oedema, suggesting the involvement of mitophagy. We also found that the PD-induced protective effects were largely reversed in Parkin−/− mice. These findings indicate that PD-induced Parkin-dependent mitophagy has a protective role in ARDS.
In conclusion, the present study provides strong evidence that PD significantly activates Parkin-dependent mitophagy to protect against mitochondria-dependent apoptosis and lung injury in ARDS (Fig. 6). Combined with the results of our previous report [20], the mitophagy-inducing function of PD warrants further exploration for ARDS therapy.

PD protects against mitochondria-dependent apoptosis by upregulation of Parkin-dependent mitophagy in acute lung injury. PD treatment activates Parkin-mediated mitophagy, which inhibits LPS-induced upregulation of Bax protein, downregulation of Bcl-2 protein, cytochrome c release and caspase-3 activation; ultimately reduces apoptosis and lung injury
References
Chen BB, Coon TA, Glasser JR, Zou C, Ellis B, Das T, et al. E3 ligase subunit Fbxo15 and PINK1 kinase regulate cardiolipin synthase 1 stability and mitochondrial function in pneumonia. Cell Rep. 2014;7:476–87.
Hiruma T, Tsuyuzaki H, Uchida K, Trapnell BC, Yamamura Y, Kusakabe Y, et al. IFN-β improves sepsis-related alveolar macrophage dysfunction and post-septic ARDS-related mortality. Am J Respir Cell Mol Biol. 2018;59:45–5.
Liu H, Liu P, Shi X, Yin D, Zhao J. NR4A2 protects cardiomyocytes against myocardial infarction injury by promoting autophagy. Cell Death Discov. 2018;4:27.
Zhu X, Huang L, Gong J, Shi C, Wang Z, Ye B, et al. NF-kappaB pathway link with ER stress-induced autophagy and apoptosis in cervical tumor cells. Cell Death Discov. 2017;3:17059.
Feng Y, Madungwe NB, Da CJC, Bopassa JC. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol. 2017;174:4329–44.
Checler F, Goiran T, Alves DCC. Presenilins at the crossroad of a functional interplay between PARK2/PARKIN and PINK1 to control mitophagy: implication for neurodegenerative diseases. Autophagy. 2017;13:2004–5.
Gao Y, Chen T, Lei X, Li Y, Dai X, Cao Y, et al. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep. 2016;14:5481–8.
Cao S, Shrestha S, Li J, Yu X, Chen J, Yan F, et al. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci Rep. 2017;7:2417.
Xiao B, Deng X, Lim G, Xie S, Zhou ZD, Lim KL, et al. Superoxide drives progression of Parkin/PINK1-dependent mitophagy following translocation of Parkin to mitochondria. Cell Death Dis. 2017;8:e3097.
Bernardini JP, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene. 2017;36:1315–27.
Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124:3987–4003.
Zhang C, Yu X, Gao J, Zhang Q, Sun S, Zhu H, et al. PINK1/Parkin-mediated mitophagy was activated against 1,4-enzoquinone-induced apoptosis in HL-60 cells. Toxicol In Vitro. 2018;50:217–24.
Shen Z, Zheng Y, Wu J, Chen Y, Wu X, Zhou Y, et al. PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window. Autophagy. 2017;13:473–85.
Aggarwal S, Mannam P, Zhang J. Differential regulation of autophagy and mitophagy in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2016;311:L433–L452.
Chang AL, Ulrich A, Suliman HB, Piantadosi CA. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med. 2015;78:179–89.
Wang HT, Lin JH, Yang CH, Haung CH, Weng CW, Maan-Yuh LA, et al. Acrolein induces mtDNA damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget. 2017;8:70406–21.
Li T, Cai S, Zeng Z, Zhang J, Gao Y, Wang X, et al. Protective effect of polydatin against burn-induced lung injury in rats. Respir Care. 2014;59:1412–21.
Zeng Z, Chen Z, Li T, Zhang J, Gao Y, Xu S, et al. Polydatin: a new therapeutic agent against multiorgan dysfunction. J Surg Res. 2015;198:192–9.
Gao Y, Zeng Z, Li T, Xu S, Wang X, Chen Z, et al. Polydatin inhibits mitochondrial dysfunction in the renal tubular epithelial cells of a rat model of sepsis-induced acute kidney injury. Anesth Analg. 2015;121:1251–60.
Li T, Liu Y, Li G, Wang X, Zeng Z, Cai S, et al. Polydatin attenuates ipopolysaccharide-induced acute lung injury in rats. Int J Clin Exp Pathol. 2014;7:8401–10.
Chen K, Dai H, Yuan J, Chen J, Lin L, Zhang W, et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018;9:105.
Gharanei M, Hussain A, Janneh O, Maddock H. Attenuation of doxorubicin-induced cardiotoxicity by mdivi-1: a mitochondrial division/mitophagy inhibitor. PLoS ONE. 2013;8:e77713.
Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic Biol Med. 2016;100:210–22.
Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W, et al. PINK1-Parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS ONE. 2015;10:e132499.
Dai H, Deng Y, Zhang J, Han H, Zhao M, Li Y, et al. PINK1/Parkin-mediated mitophagy alleviates chlorpyrifos-induced apoptosis in SH-SY5Y cells. Toxicology. 2015;334:72–80.
Jimenez RE, Kubli DA, Gustafsson AB. Autophagy and mitophagy in the myocardium: therapeutic potential and concerns. Br J Pharmacol. 2014;171:1907–16.
Lin MY, Cheng XT, Xie Y, Cai Q, Sheng ZH. Removing dysfunctional mitochondria from axons independent of mitophagy under pathophysiological conditions. Autophagy. 2017;13:1792–4.
Bian X, Teng T, Zhao H, Qin J, Qiao Z, Sun Y, et al. Zinc prevents mitochondrial superoxide generation by inducing mitophagy in the setting of hypoxia/reoxygenation in cardiac cells. Free Radic Res. 2018;52:80–91.
Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14:880–97.
Lin B, Liu Y, Li T, Zeng K, Cai S, Zeng Z, et al. Ulinastatin mediates protection against vascular hyperpermeability following hemorrhagic shock. Int J Clin Exp Pathol. 2015;8:7685–93.
Li G, Li T, Li Y, Cai S, Zhang Z, Zeng Z, et al. Ulinastatin inhibits oxidant-induced endothelial hyperpermeability and apoptotic signaling. Int J Clin Exp Pathol. 2014;7:7342–50.
Xie FJ, Zheng QQ, Qin J, Zhang LL, Han N, Mao WM. Autophagy inhibition stimulates apoptosis in oesophageal squamous cell carcinoma treated with fasudil. J Cancer. 2018;9:1050–6.
Wu Q, Gao C, Wang H, Zhang X, Li Q, Gu Z, et al. Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int J Biochem Cell Biol. 2018;94:44–55.
Zhou M, Xia ZY, Lei SQ, Leng Y, Xue R. Role of mitophagy regulated by Parkin/DJ-1 in remote ischemic postconditioning-induced mitigation of focal cerebral ischemia-reperfusion. Eur Rev Med Pharmacol Sci. 2015;19:4866–71.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant numbers: 81500066, 81670075, 81601708), the Natural Science Foundation of Hunan Province (grant numbers: 2018JJ6004, 2018JJ3015), the Joint Funds for the innovation of science and Technology of Fujian province (grant numbers: 2016Y9012), the Natural Science Foundation of Fujian Province (grant numbers: 2016J01451) and the Natural Science Foundation of Guangdong Province (grant numbers: 2016A030313561).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, T., Liu, Y., Xu, W. et al. Polydatin mediates Parkin-dependent mitophagy and protects against mitochondria-dependent apoptosis in acute respiratory distress syndrome. Lab Invest 99, 819–829 (2019). https://doi.org/10.1038/s41374-019-0191-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0191-3
This article is cited by
-
Activators of SIRT1 in the kidney and protective effects of SIRT1 during acute kidney injury (AKI) (effect of SIRT1 activators on acute kidney injury)
Clinical and Experimental Nephrology (2021)
-
Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury
Journal of Translational Medicine (2020)
-
The upshot of Polyphenolic compounds on immunity amid COVID-19 pandemic and other emerging communicable diseases: An appraisal
Natural Products and Bioprospecting (2020)
