
Abstract
The genome-wide clustered regularly interspaced short palindromic repeats (CRISPR) screen is a powerful tool used to identify therapeutic targets that can be harnessed for cancer treatment. This study aimed to assess the efficacy of genome-wide CRISPR screening to identify druggable genes associated with sorafenib-treated hepatocellular carcinoma (HCC). A genome-scale CRISPR knockout (GeCKO v2) library containing 123,411 single guide RNAs (sgRNAs) was used to identify loss-of-function mutations conferring sorafenib resistance upon HCC cells. Resistance gene screens identified SGOL1 as an indicator of prognosis of patients treated with sorafenib. Of the 19,050 genes tested, the knockout screen identified inhibition of SGOL1 expression as the most-effective genetic suppressor of sorafenib activity. Analysis of the survival of 210 patients with HCC after hepatic resection revealed that high SGOL1 expression shortened overall survival (P = 0.021). Further, matched pairs analysis of the TCGA database revealed that SGOL1 is differentially expressed. When we used a lentivirus Cas9 vector to determine the effect of targeting SGOL1 with a specific sgRNA in HCC cells, we found that SGOL1 expression was efficiently inhibited and that loss of SGOL1 was associated with sorafenib resistance. Further, loss of SGOL1 from HCC cell decreased the cytotoxicity of sorafenib in vivo. We conclude that the CRISPR screen is a powerful tool for therapeutic target analysis of sorafenib treatment and that SGOL1 serves as a druggable target for HCC treated with sorafenib and an indicator of prognosis.
Similar content being viewed by others


Background
Hepatocellular carcinoma (HCC) is one of the most common cancers, and its incidence is increasing in regions with historically low incidences, such as Europe and the United States [1, 2]. Although sorafenib and other agents are available for treating HCC, overall survival has not markedly increased since 2013 [3, 4]. Strategies that increase the efficacy of sorafenib are therefore urgently required. Many factors may contribute to the low efficacy of targeted agents, although the main reason is our limited understanding of the highly complex nature of HCC, and its phenotypic heterogeneity [5].
Genome-wide analyses and targeted loss-of-function pooled screens using the RNA-guided CRISPR (clustered regularly interspaced short palindromic repeats)-associated nuclease Cas9 in human cells reveals new physiological and pathological mechanisms in diverse biological models [6, 7]. CRISPR pooled libraries comprise thousands of plasmids, each containing a single guide RNA (sgRNA) that targets a specific gene. Moreover, the advent of next-generation sequencing (NGS) has increased our knowledge of tumor heterogeneity, thus enhancing our understanding of the complexity of HCC [5, 8].
CRISPR screening experiments require electroporation to amplify the pooled library, and NGS confirms library representation. Thus, high-throughput molecular and functional profiling of human cells is a key driver of precision medicine [9, 10]. Moreover, NGS technologies provide novel opportunities and challenges for precision medicine in the age of Big Data [11]. The United States Food and Drug Administration is contemplating NGS regulations as a part of the President’s Precision Medicine Initiative [12, 13]. For example, sorafenib is the first systemic agent used to treat advanced HCC, although outcomes are disappointing, partly because the “actionable” driver genes of HCC associated with sorafenib activity are unknown [14]. Biomarker-directed approaches to sorafenib therapy, specifically through genomic analyses, represent an example of the foundation of precision medicine [13].
To understand the phenotypic heterogeneity of HCC and to identify new biomarkers to evaluate and improve sorafenib therapy, we used Genome-scale Human GeCKO v2 sgRNA libraries comprising 123,411 sgRNAs to identify loss-of-function mutations representing sorafenib-associated druggable targets in HCC cells [15]. In screens of druggable target genes, we selected the hits of all enriched sgRNAs corresponding to essential genes. Of the ~ 19,050 genes tested, the knockout screen identified loss of SGOL1 as the most-effective genetic suppressor of sorafenib activity.
SGOL1 protects centromeric cohesin from cleavage during mitotic prophase by regulating the phosphorylation of a cohesin subunit [16]. Although SGOL1 functions in mitosis, its role in carcinogenesis and drug resistance is controversial [16]. Moreover, SGOL1 is associated with worse prognosis of patients with HCC. Here we collectively reviewed 210 patients with HCC who underwent liver resection [17]. To improve data accuracy and reliability, mRNAs expressed by HCCs were assessed using data sets provided by the Cancer Genome Atlas (TCGA) tool [18]. The sequence data of 20,531 genes of 424 patients revealed matches of the information of 50 patients. Subsequently, matched pairs analysis indicated that SGOL1 was differentially expressed.
In the present study, we evaluated whether loss of SGOL1 was associated with resistance to sorafenib in vitro and in vivo.
Materials and methods
Reagents
Sorafenib was purchased from Selleck Chemicals (Houston, TX, USA). The BCA protein assay kit, the Cell Counting Kit-8, and the Annexin V-FITC apoptosis detection kit were purchased from KeyGen Biotech (Nanjing, China). The EdU Apollo567 In Vitro Imaging Kit was purchased from Guangzhou, People’s Republic of China. DAPI and paraformaldehyde (PFA) were purchased from GuGe Biotech Co. Ltd. (Wuhan, China). Triton X-100 was purchased from ShenGong Biotech. Co. Ltd. (Shanghai, China). Methanol and ethanol were purchased from Shanghai LingFeng Chemical Reagent Co. Ltd. DMSO, Tween 20 and glycine was purchased from Sangon Biotech Co. Ltd. (Shanghai, China). Skim milk was purchased from Becton, Dickinson (San Diego, CA, USA). Primary antibodies against c-caspase-3, c-caspase-9, and c-PARP were purchased from Cell Signaling Technology (Danvers, MA, USA); GAPDH and SGOL1 were purchased from Abcam (Cambridge, MA, USA).
Cell lines, human subjects, tissue microarrays, immunohistochemistry, and follow-up
The human HCC cell lines Huh7 and SMMC-7721 were purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences, Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Cell lines were cultured in Minimum Essential Medium (MEM; Gibco-Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum.
Tissue microarray chips contained 210 biopsy samples of paired HCC tissues. The study was approved by the Research Ethics Committee (The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University; First Affiliated Hospital, School of Medicine, Zhejiang University). Written informed consent was obtained from all patients at the time of admission, authorizing the use of tissue specimens for scientific purpose. The follow-up data for all patients with HCC were reviewed. Measurement of survival rates was initiated at the time of surgery.
Genome-scale Cas9-mediated knockout screen
The Human CRISPR Knockout Pooled Library was acquired from Addgene (http://www.addgene.org/crispr/libraries/geckov2/). The lentiCRISPR vector was produced in HEK293T cells and concentrated to increase virus titer. Lentivirus packaging and purification followed the protocols of GeCKO v2 [15, 19, 20]. We first determined the concentration of sorafenib required to inhibit the proliferation of Huh7 cells. Huh7 cells were infected at a low multiplicity of infection (0.4) to ensure that most cells received the viral construct. The concentrated GECKO library (30 µl) was applied to each well containing 3 × 106 cells, yielding a transduction efficiency of 30% (270 cells per lentiCRISPR construct). Cells were selected with puromycin (1 µg/ml) 72 h after infection and then incubated for 5 days in the presence of puromycin to maximize viral integration and gene expression. The transduced Huh7 cells were split equally in two with a minimum of 3 × 106 cells per replicate, and 3 × 106 cells were cryogenically preserved for subsequent genomic DNA analysis. Two replicates were cultured in opti-MEM supplemented with 7.5 µM sorafenib, and two replicates were cultured in opti-MEM supplemented with an equal volume of DMSO (vehicle). Replicates were passaged, or fresh media was added every 3 days. Cell pellets (3 × 106) cells were prepared 14 days after drug addition. Frozen cell pellets were thawed, and genomic DNA was extracted with using a TIANamp Genomic DNA Kit, DP304 (Tiangan, China). PCR was performed following the manufacturer’s protocol. Primer sequences for amplifying lentiCRISPR sgRNAs for the first PCR were FI, TCTTGTGGAAAGGACGAAACACCG and RI, CTCTAGAACCGGTCCTGTGTTCTG. The amplicons were extracted from 2% agarose gels using an EasyPure Quick Gel Extraction Kit (Transgent, China), quantified, mixed, and then sequenced by the Novogene Bioinformatics Institute (Beijing, China).
Lentiviral Cas9-easy-vector
The sgRNAs targeting SGOL1 from the GeCKO v2 library were cloned into the lentiviral Cas9 vector (U6-sgRNA-EF1a-Cas9-FLAG-P2A-EGFP). Genechem (Shanghai, China) designed and cloned the corresponding sgRNAs into the vector [21], and a nontargeting sgRNAs (NC-sgRNA) were generated as controls. The SGOL1-sgRNA and NC-sgRNA sequences were TGTCAGCTATATGCATTGAA and CGCTTCCGCGGCCCGTTCAA, respectively. In brief, Huh7 and SMMC-7721 cells were infected with the lentivirus CRISPR construct harboring sgRNAs targeting SGOL1 or NC guides, using the original protocol for the GeCKO v2 screen. Puromycin (1 µg/ml) was added the day after infection. Selection was performed for 5 days before determining growth curves. SGOL1 expression was analyzed using western blotting. The Knockout and Mutation Detection Kit (Genechem, Shanghai, China) was used to analyze HCC cells at the indicated days postinfection. The cells transduced with SGOL1-sgRNA and NC-sgRNA were added at an appropriate concentration to 24-well or 6-well plates. Cells were cultured in MEM media with DMSO or sorafenib and counted in duplicate wells.
Western blot analysis and RT-PCR
In brief, the cells were lysed and sonicated. Protein concentrations were quantified using the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions. After denaturation, the proteins were separated using 12% SDS-PAGE, electrophoretically transferred to PVDF membranes (Bio-Rad Laboratories, Hercules, CA, USA), and incubated with blocking buffer for 2 h. After incubation at 4 °C overnight with primary antibodies (diluted 1:1000), the membranes were incubated at 37 °C for 2 h with appropriate secondary antibodies. GAPDH (diluted 1:1000) was used as a control. Blots were visualized using ECL detection reagents and quantified with Image Lab software (Bio-Rad Laboratories, Hercules, CA, USA). The reagents used and the detailed RT-PCR procedures are published [22, 23].
Cell proliferation assay
HCC cells were treated with various concentrations of sorafenib or DMSO for 24 h, 48 h, and 72 h. Cell proliferation was determined using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. The effect of sorafenib on cell viability was assessed using a colorimetric immunoassay (Cell-Light EdU Apollo567 In Vitro Imaging Kit; Ribobio, Guangzhou, China) according to the manufacturer’s instructions.
Detection of cell death
To determine the percentage of apoptotic cells after sorafenib treatment, we used the Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s protocols (KeyGen, Biotech, Nanjing, China). Huh7 and SMMC-7721 cells were seeded into six-well plates, and control and treated cells were collected after exposure to sorafenib. The HCC cells were then washed with phosphate-buffered saline (PBS), incubated with 100 µl RNase A (10 µg/ml) for 30 min, and then resuspended in 400 µl propidium iodide (PI) (50 µg/ml) for 30 min at room temperature in the dark. To analyze apoptosis after sorafenib or DMSO treatment, the cells were trypsinized, washed in cold PBS, and then pelleted. The cells were then stained using the Annexin V-FITC reaction reagent (Annexin V-FITC and PI, 5 µl each) at room temperature for 20 min in the dark and analyzed using a BD LSR II flow cytometer. The In Situ Cell Death Detection Kit (Roche, Germany) provides a nonradioactive technique for detecting and quantifying apoptosis. The TUNEL reaction preferentially labels DNA strand breaks generated during apoptosis. The experiments were performed according to the manufacturer’s instructions.
Mouse model
Animal experiments were performed in accordance with the National Institutes of Health’s guidelines. The BALB/c nu mouse (body weights 18–20 g, 6–7-week-old females) were purchased from the Zhejiang Provincial Academy of Medical Sciences (Animal Experimental Center) and maintained under defined conditions in the National Organ Key Laboratory. Cells (2 × 106) were resuspended in 100 ml of PBS and then injected subcutaneously into the flanks of nude mice (n = 28). The mice were treated with a sorafenib (30 mg/kg) and olive oil (negative control) gavage once every 2 days for 28 days. Tumor volumes and body weights were measured every 4 days for 28 days, and tumors were harvested after 28 days. Tumor volumes were calculated as follows: V (cm3) = width2 (cm2) × length (cm)/2.
Statistical analysis
Data are expressed as the mean ± standard deviation. Kaplan–Meier survival curves and log-rank tests were performed to compare SGOL1-positive and SGOL1-negative groups. A two-tailed Student's t-test and analysis of variance of multiple comparisons were used for analyses. P < 0.05 (two-sided) was considered statistically significant. Data were analyzed using SPSS19.0 software.
Results
A genome-wide screen to detect genes associated with resistance to sorafenib
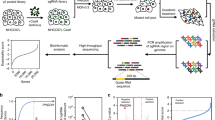
Sorafenib is a multikinase inhibitor that exhibits antiangiogenic and antitumor activities. Few studies report the use of genome-wide screens to identify druggable targets. CRISPR/Cas9 is a useful tool for genetic screening experiments, because its use of gRNAs and Cas9 can modify virtually any genetic locus. First, we determined concentration of sorafenib that killed Huh7 cells (Fig. 1a). We employed a library (Human GeCKO v2) comprising 123,411 sgRNAs corresponding to unique siRNA duplexes targeting 19,050 unique human genes (Fig. 2a). This whole-genome Cas9-sgRNA library is available from Addgene (http://www.addgene.org/crispr/libraries/geckov2/). After three treatments with sorafenib (7.5 µM) for 14 days, viable Huh7 cells were collected to determine the enrichment of sgRNAs. The sgRNA-coding regions were PCR-amplified, sequenced and subjected to NGS to determine the representation of the library (Fig. 2b). Our genome-wide CRISPR screen detected enrichment of multiple sgRNAs, suggesting that loss of their cognate genes contributed to sorafenib resistance. SGOL1 represented most hits (File S1). All log10-fold changes of the normalized counts were combined into one heat map and ranked according to the enrichment level of sorafenib/NC or NC/sorafenib (Fig. 3a). Dz-value analysis was used to generate a summary statistic by combining information corresponding to all sgRNAs for a given gene.

Sorafenib sensitivity profiles. a The proliferation of HCC cells after sorafenib treatment was assessed using the CCK-8 assay. b Western blotting and the Knockout and Mutation Detection Kit was used to evaluate if the sgRNAs efficiently knocked out SGOL1 from HCC cells. c The proliferation rates of HCC cells after treated with 5 µM or 7.5 µM sorafenib for 72 h. d EdU assays of HCC cells after incubation for 72 h with sorafenib (7.5 µM). Data are presented as the mean ± SD, *P < 0.05

Genome-scale Cas9-mediated knockout screen identifies a druggable target to enhance sorafenib treatment of hepatocellular carcinoma (HCC). a Design of an sgRNA library for genome-scale knockout of coding sequences from Huh7 cells. Huh7 cells were treated with the GeCKO v2 library to create the CRISPR library and treated with sorafenib (7.5 µM) or vehicle (DMSO). Viable Huh7 cells were collected to analyze the enrichment of sgRNAs harboring coding regions. These sgRNAs were PCR-amplified and sequenced. NGS was performed to determine the representation of the library. b Sorafenib resistance screen of Huh7 cells

Genome-scale Cas9-mediated knockout screen of Huh7 cells reveals genes whose loss confers sorafenib resistance. a The log10-fold changes of normalized counts were incorporated into one heat map and ranked according to enrichment levels from sorafenib/NC or NC/sorafenib. b Box plot demonstrating the distribution of sgRNA frequencies at different times, enrichment of specific sgRNAs as follows: day 0 (D0) Vehicle, 14 days (D14) of sorafenib treatment, candidate sgRNA. c Scatterplot demonstrating enrichment of sgRNAs after sorafenib treatment. d Identification of target genes using Dz-value analysis. e The genes were analyzed using the Gene Ontology database
The sgRNAs were ranked according to their twofold enrichment between groups. Of the 19,050 genes tested, the knockout screen identified inhibition of SGOL1 expression as the most-effective genetic suppressor of sorafenib activity (Fig. 3c, d). The hits were further analyzed using Gene Ontology (http://www.geneon-tology.org). The most highly enriched GO target was “chromosome, centromeric region” (Fig. 3e).
SGOL1 expression is associated with worse prognosis of HCC
SGOL1 protects centromeric cohesin from cleavage in mitotic prophase by regulating phosphorylation of a cohesin subunit, and evidence indicates that SGOL1 is associated with oncogenesis [16]. However, the contribution of SGOL1 to the resistance of HCC cells to sorafenib is unknown. Immunohistochemical analysis of 210 pairs of HCC samples detected SGOL1 expression in the nucleus.
There was a statistically significant correlation between SGOL1 expression and survival (Fig. 4a). Kaplan–Meier analysis revealed that patients with HCC with high SGOL1 expression experienced worse overall survival (P = 0.021; 95% confidence interval, 38.55–53.22) (Fig. 4b). SGOL1 mRNA was expressed at higher levels in HCC tumor tissues (Fig. 4c). SGOL1 protein expression was determined by western blotting in 8HCC tissues (Fig. 4d). The TCGA, which comprises a large sample of mRNA sequences, was used to assess SGOL1 expression. The TCGA database provides normalized count data of RNA sequences, including mRNA expression profiles obtained using the RNASeqV2 (level-3) system. HCC level-3 RNA sequencing data, downloaded from the TCGA database (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htmlmode = ApplyFilter&showMatrix = true&diseaseType = LIHC&tumorNormal = TN&tumorNormal = T&tumorNormal = NT), were collected using the Illumina HiSeq 2000 RNA sequencing platform (Illumina Inc., Hayward, CA, USA).The sequencing data of 20,531 genes of 424 patients revealed matched of the information of 50 patients. Subsequently, matched-pair analysis indicated that SGOL1 was differentially expressed (P < 0.05) (File S2).

Increased levels of SGOL1 indicate worse patient prognosis. a HCC samples in a tissue microarray were reacted with anti-SGOL1 monoclonal antibodies. b Survival analysis of SGOL1- negative and -positive patients (log-rank test (P = 0.021; 95% CI, 38.55–53.22). c Relative SGOL1 mRNA levels were higher in 30 pairs of HCC tissues compared with those of adjacent tissues. d Western blot analysis of SGOL1 expression in eight HCC tissues (C: cancer tissue, A: adjacent tissue). Scale bar = 100 μm
Loss of SGOL1 is associated with sorafenib resistance
We next evaluated whether SGOL1 expression was associated with sorafenib resistance. For this purpose, we used an sgRNA-expressing lentivirus to knockout SGOL1 from HCC cells. Western blotting was performed to evaluate whether SGOL1-sgRNA inhibited SGOL1 expression, (Fig. 1b). Further, the Knockout and Mutation Detection Kit (Genechem, Shanghai, China) was used to determine whether the sgRNA efficiently knocked out SGOL1 vs the NC-sgRNA control (Fig. 1b). The proliferation of HCC cells was assessed using the CCK-8 and 5-ethynyl-20-deoxyuridine (EdU) assays. Next, HCC cells expressing SGOL1-sgRNA or NC-sgRNA were treated with 5 µM or 7.5 µM of sorafenib for 72 h. CCK-8 assays and EdU assays revealed that SGOL1-sgRNA significantly decreased the cytotoxic effects of sorafenib on HCC cells (Fig. 1c, d).
Knockout of SGOL1 reduces apoptosis after sorafenib treatment
Knockdown of SGOL1 in colorectal cancer cells leads to mitotic and chromosomal instability and apoptosis [16]. We therefore investigated if knockout of SGOL1 led to apoptosis. SGOL1-sgRNA induced apoptosis of HCC cells to the same extent compared with NC-sgRNA cells (P > 0.05) (Fig. 5a). These results are consistent with those of the CRISPR screen that frequently identified SGOL1.

SGOL1 knockout from HCC cells decreases the cytotoxicity of sorafenib. a Cells expressing NC-sgRNA and SGOL1-sgRNA were treated with sorafenib (7.5 µM) for 72 h. Apoptosis rates were determined using Annexin V-FITC/PI. The data are shown as the mean ± SD from three experiments, *P < 0.05. b Western blot analysis of proteins that mediate apoptosis. Data are shown as the mean ± SD. *P < 0.05. c TUNEL and immunohistochemical analyses of apoptosis of cells expressing SGOL1-sgRNA. Data represent the mean ± SD. *P < 0.05. Scale bar = 50 μm
We next found that sorafenib treatment reduced apoptosis of SGOL1-sgRNA HCC cells. Annexin V-FITC/PI experiments showed that sorafenib-treated NC-sgRNA cells underwent apoptosis at significantly higher rates compared with those of SGOL1-depleted cells (Fig. 5a). Further, TUNEL assays revealed significantly decreased the number of apoptotic cells in SGOL1-depleted cells compared with controls (Fig. 5c). Western blotting detected proteins that mediate apoptosis, including c-caspase-9, c-caspase-3, and c-PARP, which is consistent with the Annexin V-FITC/PI and TUNEL data (Fig. 5b).
Loss of SGOL1 decreases sorafenib cytotoxicity in vivo
We next evaluated whether loss of SGOL1 from HCC decreased sorafenib cytotoxicity in vivo. Huh7 (NC-sgRNA and SGOL1-sgRNA) cells were subcutaneously inoculated into nude mice. Sorafenib was administered through gavage using a solution of olive oil. Mice were treated with the sorafenib (30 mg/kg) and olive oil (negative control) gavage once every 2 days for 28 days. The tumor masses of SGOL1-sgRNA-treated mice were significantly larger compared with those of NC-sgRNA-mice (Fig. 6a-c). The tumor sizes of NC-sgRNA–mice were significantly smaller compared with those of the SGOL1-sgRNA-mice, with no significant loss of body weight (Fig. 6e, f). Moreover, western blotting and immunohistochemical analysis detected proteins that mediate apoptosis, such as c-caspase-3 and c-PARP (Fig. 6d, g). These results are consistent with the in vitro results.

The effects of sorafenib treatment on the SGOL1-sgRNA Huh7 cells in vivo. a Huh7 cells were subcutaneously inoculated into nude mice. Mice were treated with a sorafenib (30 mg/kg) and olive oil (negative control) gavage once every 2 days for 28 days. Tumor weights (g) after the completion of treatment were significant. Tumor volumes (mm3) and body weights were measured once every 4 days. b RT-PCR analysis of relative SGOL1 mRNA expression. c The tumor masses of four groups of animals were compared. d Western blot analysis of c-caspase-3 and c-PARP expression in tumor xenograft tissues. e The tumor volumes of the nude mice were measured and calculated once every 2 days. f The body weights of nude mice were measured every 4 days. Data are shown as the mean ± SD. *P < 0.05 vs control group. g Immunohistochemical analyses of SGOL1, c-caspase-3, and c-PARP expression in tumor xenograft tissues. Scale bar = 100 μm
Discussion
An unique opportunity has arisen to deploy genome-wide CRISPR screening of agents involved in HCC to identify new molecular targets [6, 12, 24]. The current study demonstrates that CRISPR/Cas9-mediated genome editing in combination with NGS serves as a powerful tool for discovering druggable targets of sorafenib activity to guide treatment. Currently, no such clear, predictive druggable target has been identified to guide sorafenib treatment of HCC [25]. Our data show that SGOL1 may serve as a druggable target to enhance sorafenib activity, specifically because high SGOL1 expression correlated with shorter overall survival of patients with HCC after hepatectomy. To the best of our knowledge, this is the first study to utilize genome-wide CRISPR screening to identify a predictive biomarker to select patients who are most likely to benefit from sorafenib treatment [26].
SGOL1, which has a pivotal role in sister chromatid cohesion during mitosis, protects centromeric cohesion from cleavage during mitotic prophase by preventing phosphorylation of a cohesion subunit [27, 28]. Reduced SGOL1 expression leads to premature loss of sister chromatid missegregation, centromeric cohesion, and mitotic arrest [28, 29]. SGOL1 is a potential therapeutic target for HCC because of its role in cell division [30]. Here we show that SGOL1 was the most highly enriched gene detected using the CRISPR screen, indicating that knockout of SGOL1 contributed to sorafenib resistance.
In support of this possibility, we found that knockout of SGOL1, achieved using an sgRNA, induced apoptosis; however, similar results were obtained when we used the control sgRNA. The difference between these results from those of a previous study may be explained by the use of phenotypically different cell lines. As discussed above, our understanding of the basis of the marked heterogeneity of HCCs is limited. Similarly, the mechanism through which SGOL1 contributes to oncogenesis is unknown.
To confirm the reliability and efficacy of genome-wide CRISPR screening, we first used Dz-value analysis to evaluate sgRNAs that targeted the top five candidate genes. These genes, which yielded new testable hypotheses regarding the mechanism of sorafenib resistance, include SGOL1, v-set and transmembrane domain containing 2 like, cyclin-dependent kinase inhibitor 1C, testis specific serine kinase 4, and ubiquitin specific peptidase 6 (File S1). Together with the results of our experiments presented here employing an HCC cell line and a mouse xenograft model of HCC, we conclude that SGOL1 will serve as a new druggable target of sorafenib resistance of HCC. To exploit this possibility will require further research to identify the details of the molecular mechanisms involved. For example, distinct responses to mitotic checkpoint activation among normal and HCC cells may facilitate efforts to selectively target HCC cells with sorafenib, potentially avoiding its adverse effects.
Together, our findings show that the CRISPR screen serves as a powerful tool for the discovery of therapeutic targets to enhance sorafenib treatment. Our experience using CRISPR/Cas9 may serve as a new strategy for enhancing our knowledge of the phenotypic heterogeneity of HCC. Our data suggest that SGOL1 expression will serve as an indicator of a patient’s prognosis. The high differential expression of SGOL1 in HCC cells was associated with an increased risk of poor prognosis. Thus, selectively targeting SGOL1 in patients with HCC who are treated with sorafenib may increase survival by months, if not years.
Sorafenib is the only systemic drug approved for patients with advanced HCC. Unfortunately, sorafenib treatment increases patients’ survival by only a few months, and new therapeutic targets are therefore urgently required. However, no definitive predictive biomarkers are available to guide sorafenib treatment of advanced HCC. Our present study demonstrates that CRISPR/Cas9-mediated genome editing in combination with NGS serves as a powerful tool for discovering biomarkers to guide treatment with sorafenib. Our data support the conclusion that SGOL1 is a druggable target that may enhance the efficacy of sorafenib treatment. Further, high levels of SGOL1 are associated with shorter overall survival of patients with HCC after hepatectomy. To our knowledge, this study is the first use of genome-wide CRISPR screens to identify a predictive biomarker that can be used to select patients who are most likely to benefit from sorafenib treatment.
References
Cheng N, Li Y, Han ZG. Argonaute2 promotes tumor metastasis by way of up-regulating focal adhesion kinase expression in hepatocellular carcinoma. Hepatology. 2013;57:1906–18.
Han Y, Chen Z, Yang Y, et al. Human CD14+CTLA-4+regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59:567–79.
Abou-Alfa GK. Sorafenib use in hepatocellular carcinoma: more questions than answers. Hepatology. 2014;60:15–18.
Camma C, Cabibbo G, Petta S, et al. Cost-effectiveness of sorafenib treatment in field practice for patients with hepatocellular carcinoma. Hepatology. 2013;57:1046–54.
Chan SL, Wong AM, Lee K, et al. Personalized therapy for hepatocellular carcinoma: where are we now? Cancer Treat Rev. 2016;45:77–86.
Xue HY, Ji LJ, Gao AM, et al. CRISPR-Cas9 for medical genetic screens: applications and future perspectives. J Med Genet. 2016;53:91–97.
Savic N, Schwank G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl Res. 2016;168:15–21.
Prum RO, Berv JS, Dornburg A, et al. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature. 2015;526:569–73.
Lyman GH, Moses HL. Biomarker tests for molecularly targeted therapies--the key to unlocking precision medicine. N Engl J Med. 2016;375:4–6.
Bonham VL, Callier SL, Royal CD. Will precision medicine move us beyond race? N Engl J Med. 2016;374:2003–5.
Jameson JL, Longo DL. Precision medicine--personalized, problematic, and promising. N Engl J Med. 2015;372:2229–34.
Vicini P, Fields O, Lai E, et al. Precision medicine in the age of big data: the present and future role of large-scale unbiased sequencing in drug discovery and development. Clin Pharmacol Ther. 2016;99:198–207.
Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5.
Gores GJ. Decade in review-hepatocellular carcinoma: HCC-subtypes, stratification and sorafenib. Nat Rev Gastroenterol Hepatol. 2014;11:645–7.
Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–4.
Matsuura S, Kahyo T, Shinmura K, et al. SGOL1 variant B induces abnormal mitosis and resistance to taxane in non-small cell lung cancers. Sci Rep. 2013;3:3012.
Xu X, Lu D, Ling Q, et al. Liver transplantation for hepatocellular carcinoma beyond the Milan criteria. Gut. 2016;65:1035–41.
Liu Z, Chen P, Gao H, et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. 2014;26:106–20.
Steinhart Z, Pavlovic Z, Chandrashekhar M, et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat Med. 2017;23:60–68.
Shalem O, Sanjana NE, Hartenian E, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87.
Gao Y, Wang Z, Hao Q, et al. Loss of ERalpha induces amoeboid-like migration of breast cancer cells by downregulating vinculin. Nat Commun. 2017;8:14483.
Ling S, Xie H, Yang F, et al. Metformin potentiates the effect of arsenic trioxide suppressing intrahepatic cholangiocarcinoma: roles of p38 MAPK, ERK3, and mTORC1. J Hematol Oncol. 2017;10:59.
Xiao H, Cheng S, Tong R, et al. BAG3 regulates epithelial-mesenchymal transition and angiogenesis in human hepatocellular carcinoma. Lab Invest. 2014;94:252–61.
Zhou Y, Zhu S, Cai C, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–91.
Sohn W, Paik YH, Cho JY, et al. Sorafenib therapy for hepatocellular carcinoma with extrahepatic spread: treatment outcome and prognostic factors. J Hepatol. 2015;62:1112–21.
Colagrande S, Inghilesi AL, Aburas S, et al. Challenges of advanced hepatocellular carcinoma. World J Gastroenterol. 2016;22:7645–59.
Kahyo T, Iwaizumi M, Shinmura K, et al. A novel tumor-derived SGOL1 variant causes abnormal mitosis and unstable chromatid cohesion. Oncogene. 2011;30:4453–63.
Chetaille P, Preuss C, Burkhard S, et al. Mutations in SGOL1 cause a novel cohesinopathy affecting heart and gut rhythm. Nat Genet. 2014;46:1245–9.
Kim MS, An CH, Yoo NJ, et al. Frameshift mutations of chromosome cohesion-related genes SGOL1 and PDS5B in gastric and colorectal cancers with high microsatellite instability. Hum Pathol. 2013;44:2234–40.
Wang LH, Yen CJ, Li TN, et al. Sgo1 is a potential therapeutic target for hepatocellular carcinoma. Oncotarget. 2015;6:2023–33.
Acknowledgements
We are grateful to Xiao-Hu Zhou and Xiao-Lei Zhang for technical assistance and helpful suggestions.
Funding
This work was supported by Innovative Research Groups of the National Natural Science Foundation of China (No. 81421062), the Major program of National Natural Science Foundation of China (No.91542205), the National S&T Major Project of China (No. 2012ZX10002017), Zhejiang Provincial Natural Science Foundation of China (LY18H160046), and Zhejiang Medical Science Foundation (2018KY532).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Declarations
Ethics approval and consent to participate. The use of nude mice in this study was approved by the Medical Ethics Committee of the First Affiliated Hospital of Zhejiang University. The informed consent of the HCC samples was obtained from all patients.
These authors contributed equally: Weijian Sun, Bin He, and Beng Yang.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sun, W., He, B., Yang, B. et al. Genome-wide CRISPR screen reveals SGOL1 as a druggable target of sorafenib-treated hepatocellular carcinoma. Lab Invest 98, 734–744 (2018). https://doi.org/10.1038/s41374-018-0027-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0027-6
This article is cited by
-
Prognosis and biological function of SGOL1 in clear cell renal cell carcinoma: a multiomics analysis
BMC Medical Genomics (2024)
-
A review on CRISPR/Cas: a versatile tool for cancer screening, diagnosis, and clinic treatment
Functional & Integrative Genomics (2023)
-
CRISPR/Cas9 gene editing: a new approach for overcoming drug resistance in cancer
Cellular & Molecular Biology Letters (2022)
-
SGOL2 is a novel prognostic marker and fosters disease progression via a MAD2-mediated pathway in hepatocellular carcinoma
Biomarker Research (2022)
-
miR-15a and miR-20b sensitize hepatocellular carcinoma cells to sorafenib through repressing CDC37L1 and consequent PPIA downregulation
Cell Death Discovery (2022)
