
Abstract
The discovery of mutations in genes encoding the metabolic enzymes isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH), and fumarate hydratase (FH) has expanded our understanding not only of altered metabolic pathways but also epigenetic dysregulation in cancer. IDH1/2 mutations occur in enchondromas and chondrosarcomas in patients with the non-hereditary enchondromatosis syndromes Ollier disease and Maffucci syndrome and in sporadic tumors. IDH1/2 mutations result in excess production of the oncometabolite (d)-2-hydroxyglutarate. In contrast, SDH and FH act as tumor suppressors and genomic inactivation results in succinate and fumarate accumulation, respectively. SDH deficiency may result from germline SDHA, SDHB, SDHC, or SDHD mutations and is found in autosomal-dominant familial paraganglioma/pheochromocytoma and Carney-Stratakis syndrome, describing the combination of paraganglioma and gastrointestinal stromal tumor (GIST). In contrast, patients with the non-hereditary Carney triad, including paraganglioma, GIST, and pulmonary chondroma, usually lack germline SDH mutations and instead show epigenetic SDH complex inactivation through SDHC promoter methylation. Inactivating FH germline mutations are found in patients with hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome comprising benign cutaneous/uterine leiomyomas and renal cell carcinoma. Mutant IDH, SDH, and FH share common inhibition of α-ketoglutarate-dependent oxygenases such as the TET family of 5-methylcytosine hydroxylases preventing DNA demethylation, and Jumonji domain histone demethylases increasing histone methylation, which together inhibit cell differentiation. Ongoing studies aim to better characterize these complex alterations in cancer, the different clinical phenotypes, and variable penetrance of inherited and sporadic cancer predisposition syndromes. A better understanding of the roles of metabolic enzymes in cancer may foster the development of therapies that specifically target functional alterations in tumor cells in the future. Here, the physiologic functions of these metabolic enzymes, the mutational spectrum, and associated functional alterations will be discussed, with a focus on mesenchymal tumor predisposition syndromes.
Similar content being viewed by others


The role of metabolic enzymes in cancer
Metabolic alterations are regarded as one of the hallmarks of cancer [1] and include the phenomenon of “aerobic glycolysis”, which was first described by Otto Warburg in the 1920s and termed the “Warburg effect” [2]. It has been shown that even in the presence of oxygen, cancer cells switch from generating ATP by the highly energy-efficient process of oxidative phosphorylation to the much less efficient process of glycolysis [3, 4]. However, cancer metabolism may also be affected by various other mechanisms, such as dysregulated signaling through activated oncogenes (e.g., RAS, MYC) and mutant tumor suppressors (e.g., TP53) [1]. To date, it is still not entirely clear whether aerobic glycolysis is a true cause or rather a consequence of cancer.
The metabolic enzymes isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH), and fumarate hydratase (FH) play important roles in the tricarboxylic acid cycle (also known as the citric acid cycle or Krebs cycle). The discovery that mutations in the encoding genes can lead to cancer development offers the opportunity to further characterize the complex regulatory mechanisms of energy metabolism in cancer. Further, the roles of these enzymes beyond regulation of metabolic pathways provide important insights into epigenetic changes as potential drivers in cancer.
Change-of-function IDH mutations occur during post-zygotic divisions and lead to somatic mosaicism. They are associated with the development of multiple enchondromas in non-hereditary Ollier disease and Maffucci syndrome, which may transform to chondrosarcoma in a subset of cases [5, 6]. SDH deficiency resulting from inactivating germline mutations of the SDH subunits SDHA, SDHB, SDHC, or SDHD is associated with hereditary paraganglioma and pheochromocytoma as well as Carney-Stratakis syndrome, the latter comprising gastrointestinal stromal tumor (GIST) and paraganglioma [7, 8]. Finally, inactivating germline mutations of FH are associated with the hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome, which includes benign cutaneous and uterine leiomyomas and renal cell carcinoma [9].
Here, mutations in these metabolic enzymes, their impact on metabolic homeostasis, and the associated tumor syndromes will be discussed after a brief overview of their physiologic function in the tricarboxylic acid cycle. Their biologic roles in cancer development will be reviewed, with a focus on mesenchymal tumors, as well as opportunities for therapeutic targeting and future studies.
Physiologic roles of IDH, SDH, and FH in the tricarboxylic acid cycle
First described by Hans Adolf Krebs in 1937 [10], the tricarboxylic acid cycle is the primary metabolic pathway for all aerobic processes in animal tissues [11]. It involves a series of reactions (Fig. 1) that generate essential substrates as building blocks for biosynthesis while (indirectly) utilizing about two-thirds of the total consumed oxygen and generating about two-thirds of the total energy [11]. The tricarboxylic acid cycle is located within mitochondria, represents the final common pathway for the oxidation of carbohydrate, protein, and lipids, and plays important roles in gluconeogenesis, transamination, deamination, and lipogenesis [11].

The tricarboxylic acid cycle. Shown are the biochemical reactions catalyzed by isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH), and fumarate hydratase (FH) and oncometabolites resulting from the respective mutations. NAD indicates nicotinamide adenine dinucleotide, Q ubiquinone, and QH2 ubiquinol
IDH isoforms are crucial enzymes for the incorporation of glucose and fatty acid carbons into the tricarboxylic acid cycle. The homodimeric NADP+-dependent enzymes IDH1 and IDH2 reversibly catalyze the oxidative decarboxylation of isocitrate to produce α-ketoglutarate, NADPH, and CO2 [4, 12]. SDH is an enzymatic complex composed of the SDHA, SDHB, SDHC, and SDHD subunits embedded in the inner mitochondrial membrane and is involved not only in the tricarboxylic acid cycle but also in the electron transport chain (as complex II). SDH catalyzes the reversible oxidation of succinate to fumarate, using the electrons generated through reduction of ubiquinone to ubiquinol in the electron transport chain [12]. Finally, FH (which has both mitochondrial and cytosolic isoforms) catalyzes the reversible hydration of fumarate to (l)-malate [12].
IDH mutations lead to changes in enzyme function and generation of the oncometabolite (d)-2-hydroxyglutarate
Mammals express three IDH isoforms [13] with distinct intracellular location: IDH1 and IDH2 isoforms are NADP+-dependent enzymes that reversibly catalyze the decarboxylation of isocitrate to yield α-ketoglutarate, NADPH, and CO2, whereas IDH3 is a structurally unrelated heterotetrameric NAD+-dependent enzyme that irreversibly decarboxylates isocitrate and produces α-ketoglutarate, NADH, and CO2 [4, 12]. Whereas IDH1 is located in the cytoplasm and peroxisomes, IDH2 and IDH3 are located in the mitochondrial matrix [4].
Among IDH, SDH, and FH, genetic alterations of IDH1/2 are most frequent: mutations have been observed in tumors of various origins, such as glioma (80–90% of cases) [14], acute myeloid leukemia (AML) (~20 of cases) [15, 16], sinonasal undifferentiated carcinoma (55% of cases) [17], cholangiocarcinoma (10–20% of cases) [18], chondrosarcoma (50–70% of cases), and spindle cell hemangioma (70%) [19, 20]. Mutations in the IDH3 isoform have not been reported in cancer. It has been shown that the prognostic significance of IDH1/2 mutations varies in between different tumor types [4]: for instance, in glioblastoma, IDH1/2 mutations are associated with favorable outcome [21,22,23], whereas in AML, cholangiocarcinoma and chondrosarcoma, the prognostic significance of IDH1/2 mutations is less clear [24,25,26]. In myelodysplastic syndromes and myeloproliferative neoplasms, IDH1/2 mutations confer a poor prognosis [4].
Of note, the spectrum of IDH1 and IDH2 mutations varies among the different tumor types: for instance, in grade II/III gliomas and glioblastomas, the most common IDH1 mutation is p.R132H (85–90% of IDH mutations); IDH1 mutations are much more common than IDH2 mutations. Cholangiocarcinoma shows a predominance of IDH1 over IDH2 mutations (ratio 10:1), with the IDH1 mutation p.R132C as the most common alteration (50–60% of IDH mutations). In AML, the p.R140Q mutation in IDH2 is the most frequent alteration (30–50% of IDH mutations), with IDH1 and IDH2 mutations occurring at a ratio of 1:1–2. Chondrosarcoma harbors a spectrum of IDH mutations similar to cholangiocarcinoma, with predominance of IDH1 over IDH2 mutations (ratio 20:1) and p.R132C representing the most common amino acid exchange (40–50% of IDH mutations). Thus, the use of the R132H IDH1 mutation specific antibody, commonly applied by pathologists in glioma diagnostics [27], is not useful for the other tumor types in which other IDH1 or IDH2 mutations prevail.
Chondrosarcoma is the second most frequent primary malignant bone neoplasm [28]. During progression, chondrosarcomas accumulate genetic alterations that include dysregulation of the cell cycle and hedgehog signaling pathway as well as genetic alterations in COL2A1, NRAS, and YEATS2 [29,30,31,32,33]. Secondary chondrosarcomas may also arise in enchondromas.
The non-hereditary enchondromatosis syndromes Ollier disease and Maffucci syndrome are rare, usually show unilateral involvement, and are associated with somatic mosaic heterozygous IDH1 or IDH2 mutations (Fig. 2 and Table 1) [5]. Ollier disease comprises multiple enchondromas, and occasionally gliomas or juvenile granulosa cell tumors [34]. Maffucci syndrome includes enchondromatosis and spindle cell hemangioma, but patients may also develop chondrosarcoma, angiosarcoma, and occasionally other tumors such as astrocytoma, pituitary adenoma, juvenile granulosa cell tumor, and pancreatic adenocarcinoma [28]. The risk of developing secondary chondrosarcoma is dependent on the location of the tumors and estimated at ~40% [35]. Approximately 40–90% of tumors in these cancer predisposition syndromes harbor mutations of Arg132 of IDH1 or, less frequently, Arg172 of IDH2 [5, 6].

Tumors characterized by isocitrate dehydrogenase (IDH) deficiency. A female patient with Maffucci syndrome developed multiple enchondromas of the distal extremities (a x-ray image of the right hand performed at age 25), histologically characterized by a benign cartilaginous proliferation (b). At age 34, CT imaging (c, coronal (top) and frontal (bottom) views) identified the presence of a chondrosarcoma adjacent to the right patella (c, arrows), measuring up to 21.6 cm in greatest dimension and histologically displaying chondrosarcoma (d) with moderate cytologic atypia. Targeted sequencing revealed a heterozygous IDH1 p.R132C (c.394C>A) missense mutation (e allele fraction 31%)
In contrast to SDH and FH, which harbor loss-of-function mutations and act as bona fide tumor suppressor genes (see below), IDH1/2 mutations confer a gain of function and change the catalytic activity of the enzyme (Fig. 1): mutant IDH1/2 catalyzes the reduction of α-ketoglutarate and results in high-level production of the oncometabolite (d)-2-hydroxyglutarate [36,37,38]. In normal cells, (d)-2-hydroxyglutarate levels are very low and tightly regulated [4]. Characterizing the oncogenic properties of (d)-2-hydroxyglutarate is an area of active investigation: it has been hypothesized that (d)-2-hydroxyglutarate may promote cellular transformation by altering the redox state of cells or leading to metabolic and epigenetic changes, in particular affecting tumor suppressor enzymes that depend on the structurally related α-ketoglutarate as cosubstrate [4]. (d)-2-hydroxyglutarate inhibits α-ketoglutarate-dependent oxygenases such the TET family of 5-methlycytosine (5mC) hydroxylases (see below) [39, 40]. Also, loss of physiologic IDH activity may result in changes in mitochondrial function and promote the metabolic switch to glycolysis [4]. Experimental treatment of mesenchymal stem cells with (d)-2-hydroxyglutarate or introduction of an IDH mutation inhibits osteogenic differentiation and stimulates chondrogenic differentiation—providing a possible explanation for the development of enchondromas during bone development [41, 42].
Interestingly, preservation of the wild-type IDH1 allele seems to be necessary to allow for formation of heterodimers with mutant IDH1 and provide mutant IDH1 with (cytoplasmic) α-ketoglutarate as a substrate for generating (d)-2-hydroxyglutarate [4]. Accordingly, gliomas with homozygous IDH1 mutations show much lower levels of oncogenic (d)-2-hydroxyglutarate than those with heterozygous mutations [43], and tumors associated with Ollier disease and Maffucci syndrome generally harbor heterozygous (rather than homozygous) IDH mutations [5].
Although IDH mutations have been shown to be no longer essential to promote tumor growth in high-grade chondrosarcoma [44, 45], the detection of canonical IDH mutations may be of diagnostic value, for instance, when chondroblastic osteosarcoma enters the differential diagnosis. IDH mutations have been reported in 87% of enchondromas in patients with Ollier disease, 77% of tumors in patients with Maffucci syndrome, 86% of secondary central chondrosarcoma, 38–70% of primary central chondrosarcoma, 15% of periosteal chondrosarcoma, and 54% of dedifferentiated chondrosarcoma [5, 6, 19, 46], but are absent in peripheral chondrosarcoma, osteosarcoma, and chordoma [5, 19, 47, 48].
Identical IDH mutations in multiple tumors have been identified in 88% of patients with Ollier disease and Maffucci syndrome [5].
Inactivating SDH mutations lead to accumulation of succinate
Loss-of-function alterations of the SDH complex, also representing the mitochondrial complex II, can be observed in familial paraganglioma/pheochromocytoma [7] with autosomal-dominant inheritance (Fig. 3 and Table 1), the autosomal-dominant Carney-Stratakis syndrome (GIST and paraganglioma) [49, 50], and the non-hereditary Carney triad [51] (GIST, paraganglioma, and pulmonary chondroma). The SDH complex is composed of proteins encoded by SDHA, SDHB, SDHC, and SDHD, and loss of function of any of these four components leads to complex inactivation and loss of SDHB expression detectable by immunohistochemistry (Fig. 3) [8].

Tumors characterized by succinate dehydrogenase (SDH) deficiency. A paraganglioma arising in the retroperitoneum (a) showing loss of SDHB expression (b) and retained SDHA expression (c) in tumor cells (admixed small vessels serve as an internal control). A gastrointestinal stromal tumor (GIST) arising in the stomach, showing the typical multinodular architecture of SDH-deficient GIST (d). Another example of an SDH-deficient GIST with loss of SDHB expression (e) and retained SDHA expression (f) in tumor cells. This tumor harbored a homozygous SDHC p.R133* (c.397C>T) nonsense mutation (f, inset, allele fraction 91%)
Paraganglioma and pheochromocytoma are rare tumors that mostly affect middle-aged adults between the third and fourth decades. While the majority of these tumors behaves in a benign fashion, the excessive production of catecholamines or local mass effect may result in high morbidity and mortality in a subset of patients.
Familial paragangliomas were first described in 1933 [52]. Up to 30% of paragangliomas and pheochromocytomas harbor germline mutations in predisposing genes [53], with SDH subunit mutations being most common, followed by mutations in VHL (4–10% of cases), RET (1–5% of cases), and NF1 (1–5% of cases) [54,55,56]. More recently, germline mutations in TMEM127 [57] and MAX [58] have been identified. Germline SDH mutations are found in ~10% of apparently sporadic and >80% of familial paragangliomas/pheochromocytomas, respectively [55, 59]. Inactivating mutations in SDHA [56], SDHB [60], SDHC [61], SDHD [62], and SDHAF2 [63] follow an autosomal-dominant inheritance pattern (Table 1). SDHAF2 encodes a mitochondrial protein that is required for the flavination (i.e., enzymatic activation) of SDHA [63]. The penetrance of disease in these paraganglioma syndromes appears to be highly variable [64]. For instance, paraganglioma syndrome 1 (caused by SDHD germline mutation) and 2 (SDHAF2) are notable for high penetrance, multifocality, and parent-of-origin inheritance, with disease usually manifesting in cases of paternal origin [64]. Paraganglioma syndrome 4 (SDHB) confers increased risk of malignancy, whereas paraganglioma syndromes 3 (SDHC) and 5 (SDHA) are infrequent and show lower penetrance [64]. It remains to be determined why mutations affecting one enzymatic complex may lead to such variable clinical presentations [64].
Rarely, germline mutations in FH have been reported to predispose to malignant paraganglioma/pheochromocytoma, pointing towards a (partial) functional overlap with SDH deficiency [65,66,67].
Also, an association of germline SDH mutations and development of renal cell carcinoma has been reported: [68, 69] SDH deficiency has been shown to impair oxidative phosphorylation which renders SDH-deficient renal cell carcinoma dependent on aerobic glycolysis [68, 70]. SDH-deficient renal cell carcinomas are usually indolent tumors with a predilection for younger patients [71] and distinct morphologic features that include a predominantly solid architecture, uniform eosinophilic tumor cells with vacuolated or flocculent cytoplasmic inclusions and admixed mast cells [72]. A morphologic overlap with FH-deficient renal cell carcinoma has been described for a subset of cases [73].
In both paraganglioma and GIST, loss of SDHA expression predicts germline SDHA mutation [74,75,76]. While a combination of GIST and paraganglioma (Carney-Stratakis syndrome) seems to be rarely associated with SDHA germline mutation [74], few such cases have been observed. Carney-Stratakis syndrome is rarely associated with pituitary adenomas [77] and renal cell carcinoma [78].
SDH-deficient GISTs are characterized by several features that help distinguish these tumors from conventional KIT/PDGFRA-mutant GISTs: they virtually always arise in the stomach, show epithelioid or mixed epithelioid and spindle cell morphology, and a characteristic multinodular or plexiform growth pattern that facilitates their recognition on conventional hematoxylin and eosin-stained slides [79].
SDH-deficient GISTs show either inactivating mutations in genes encoding the SDH subunits or alternate SDHC promoter methylation causing epigenetic inactivation (Table 1) [80, 81]. Patients with Carney-Stratakis syndrome have been reported to harbor germline mutations in SDHA, SDHB, SDHC, and SDHD, whereas Carney triad is a non-hereditary condition with a predilection for females, and patients only rarely (<10%) harbor SDH subunit germline mutations [82]. Carney triad can be complete, comprising GIST, paraganglioma, and pulmonary chondroma, or incomplete with only one or two of these three manifestations.
SDH-deficient GISTs show loss of SDHB expression by immunohistochemistry: GISTs with SDHB, SDHC, SDHD mutation or SDHC promoter methylation show retained SDHA expression, whereas SDHA-mutant cases can be identified by additional SDHA loss.
Although SDH-deficient GISTs show expression of activated KIT [8], the mechanism of KIT activation remains unclear. They lack the canonical chromosomal alterations observed in KIT/PDGFRA/NF1-mutant GISTs (i.e., loss of 14q, 22q, 1p, and 15q) and instead, may show 1q deletion presumably involving the SDHC locus [50, 51]. SDH-deficient GISTs show a propensity for multifocality and lymph node metastasis, but usually follow an indolent clinical course. As demonstrated recently, risk stratification systems initially established for KIT/PDGFRA-mutant GISTs fail to predict disease progression in SDH-deficient GISTs [83].
SDH complex inactivating mutations result in accumulation of the substrate succinate (Fig. 1) which has been shown to inhibit various α-ketoglutarate-dependent dioxygenases, including the TET family of 5mC hydroxylases [84,85,86], leading to decreased hydroxylation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and subsequent decrease in 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) formation, thereby establishing a hypermethylation program (see below) [81, 87]. At the same time, levels of fumarate and malate decrease. 5hmC expression has been shown to be low to absent in SDH-deficient GISTs by immunohistochemistry [88], which is similar to observations made in other SDH- and FH-deficient tumors, corresponding to genome-wide hypermethylation [89].
Inactivating FH mutations lead to accumulation of fumarate
Mutations in FH lead to inactivation of the enzyme and subsequent accumulation of the substrate fumarate and also, to a lesser degree, succinate. High levels of fumarate cause changes in metabolism and epigenetic regulation, similar to the effects of IDH and SDH mutations (see below). By immunohistochemistry, loss of 5hmC and increased H3K9me3 levels can be detected in FH-mutant tumor cells [89].
FH germline mutations are found in the hereditary HLRCC cancer predisposition syndrome comprising hereditary cutaneous and uterine leiomyomas and renal cell carcinoma (i.e., mostly “type 2” papillary renal cell carcinoma) which follows autosomal-dominant inheritance and shows incomplete penetrance, while somatic FH mutations can be found in a small subset of uterine smooth muscle tumors (Fig. 4 and Table 1). Patients with HLRCC develop cutaneous leiomyomas in ∼76% of cases, uterine leiomyomas in almost all affected female individuals, and renal cell carcinoma in 10–16% of cases, with a median age of detection of 44 years [90].

Tumors characterized by fumarate hydratase (FH) deficiency. A renal cell carcinoma with a papillary growth pattern (a) exhibiting prominent nucleoli surrounded by a perinucleolar halo (a, inset). This tumor was shown to have a homozygous deletion of FH at 1q43 (b, arrow), leading to loss of FH expression in tumor cells (c). A pilar leiomyoma (d) showing the typical nuclear features (e, arrows) and loss of FH expression (f); endothelial cells and inflammatory cells serve as internal controls. Case courtesy of Dr. Michelle Hirsch, Department of Pathology, Brigham and Women’s Hospital
In an unselected cohort of smooth muscle tumors, non-atypical uterine leiomyomas showed FH deficiency by immunohistochemistry in 1.6%, cellular leiomyomas in 1.8%, atypical leiomyomas in 37.3%, and none of the leiomyosarcomas tested, whereas the rate of HLRCC was very low in patients with FH-deficient tumors [91]. The median age of patients with FH-deficient uterine leiomyomas was 38 years [91]. HLRCC-associated leiomyomas and renal cell carcinomas are characterized by eosinophilic cytoplasmic inclusions, prominent eosinophilic nucleoli, and characteristic perinucleolar halos (Fig. 4) [92]. Furthermore, characteristic hemangiopericytoma-like vessels have been described in FH-deficient uterine smooth muscle tumors [93]. Loss of FH expression is detectable by immunohistochemistry with relatively high specificity but only moderate sensitivity. The accumulation of fumarate also induces aberrant succination of proteins and positive staining for (S)-2-succinocysteine (2-SC) increases the diagnostic sensitivity and can be used as an adjunct biomarker to detect FH deficiency [89, 92, 94,95,96]. However, the latter marker is not commercially available.
IDH, SDH, and FH mutations inhibit TET enzymes and lead to genome-wide hypermethylation
Beyond complex changes in metabolism with a shift in substrate dependencies, IDH-, SDH-, and FH-mutant tumors share similar mechanisms of dysregulated DNA methylation and histone modification (Fig. 5): excess (d)-2-hydroxyglutarate resulting from IDH1/2 mutations, as well as accumulation of succinate in SDH-deficient tumors [84, 85], and fumarate in FH-deficient tumors [85] inhibit TET family enzymes which results in decreased hydroxylation of 5mC to 5hmC and also a subsequent decrease in 5fC and 5caC generation by TET. It has been demonstrated that inhibition of TET2 leads to genome-wide hypermethylation and global changes in gene transcription [4, 89, 97]. Accordingly, IDH1-mutant cartilaginous tumors show a hypermethylation phenotype [5]. SDH-deficient GISTs—including GISTs with SDHx subunit mutations and SDHC promoter methylation—share a common genome-wide hypermethylation phenotype, as the unifying oncogenic mechanism [81]. Methylation profiling confirmed that the epigenetic alterations observed in SDH-deficient GIST are comparable with those in IDH-mutant gliomas [97], further highlighting the role of these enzymes for epigenetic maintenance. Intracellular succinate accumulation in SDH-deficient GISTs has been shown to lead to TET inhibition and loss of 5hmC (see below) [97]. Likewise, accumulation of fumarate was shown to induce hypermethylation of DNA [98], confirming that this group of tumors with genetic aberrations in metabolic enzymes share a common mechanism of tumorigenesis through epigenetic changes including hypermethylation.

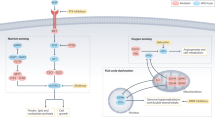
Simplified overview of proposed consequences of IDH1/2, SDH, and FH mutations for cancer development. While IDH1/2 mutations lead to functional alterations in enzyme activity and increased production of the oncometabolite (d)-2-hydroxyglutarate, SDH and FH mutations lead to accumulation of succinate and fumarate, respectively. These inhibit TET family of 5-hydroxymethylcytosine (5mC)-hydroxylases and Jumonji domain histone demethylases leading to genome-wide hypermethylation and alterations in gene transcription with a negative impact on differentiation. In addition, inhibition of hypoxia-inducible factor (HIF) prolyl hydroxylases may lead to activation in HIF-1α signaling inducing pseudohypoxia
In addition to DNA hypermethylation, (d)-2-hydroxyglutarate also inhibits other α-ketoglutarate-dependent oxygenases such as the Jumonji domain histone demethylases (JmjC), thereby increasing histone methylation [99,100,101]. Indeed, increased methylation of H3K9me3 was shown in SDH- and FH-deficient tumors [89], while this was less clear for IDH mutant cartilaginous tumors [26]. Taken together, mutations in metabolic enzymes cause epigenetic changes including both DNA hypermethylation and altered histone methylation which are shown to affect gene expression and to inhibit differentiation [40,41,42, 99].
Moreover, FH inhibition, together with accumulated fumarate and—to a lesser degree—succinate have been shown to cause intracellular hypoxia-inducible factor (HIF) accumulation and stabilization in part by inhibiting HIF prolyl hydroxylase [102]. An increase in reactive oxygen species and inhibition of HIF-α prolyl hydroxylases in the cytosol, leading to HIF-1α stabilization and activation, has been observed in SDH- and FH-deficient tumors [4, 103]. However, the concept of pseudohypoxia is still controversial and data are limited: while it has been proposed that HIF has tumor suppressor functions in myeloid leukemia, it is not clear whether the HIF response to hypoxia is impaired in IDH-mutant tumors in vivo [4], and additional studies are needed to better characterize the functional effects of dysregulated HIF signaling in mesenchymal tumors with mutations in IDH, SDH, and FH.
Therapeutic targeting of cancers driven by IDH, SDH, and FH mutations
Selectively targeting mutant IDH, SDH, and FH while preserving wild-type enzyme function in nonneoplastic cells is challenging; the biologic context in which the mutation occurs further determines sensitivity to treatment. Table 2 summarizes selected ongoing clinical trials for the treatment of IDH-, SDH-, and FH-mutant solid tumors.
In the past few years, mutant IDH inhibitors have been developed to suppress the production of (d)-2-hydroxyglutarate and have been tested in in vitro and in vivo models [104,105,106]. In IDH1-mutant chondrosarcoma cell lines, the mutant IDH1 inhibitor AGI-5198 was shown to decrease (d)-2-hydroxyglutarate levels resulting in moderately decreased viability [44, 45]. However, proliferation and migration were not affected, global gene expression, CpG island methylation as well as histone H3K4, -9, and -27 trimethylation levels remained unchanged, indicating that mutant IDH1 inhibition alone may not be sufficient in chondrosarcomas, which seem to no longer rely on these mutations, emphasizing the need for development of alternative or combinatorial strategies, that may exploit the epigenetic changes or the metabolic vulnerability of IDH mutant chondrosarcomas [44].
The oral mutant IDH2-inhibitor enasidenib has recently been approved by the Food and Drug Administration (FDA) for the treatment of relapsed or refractory AML in patients with detectable IDH2 mutation by FDA-approved genetic testing [107]. It remains to be seen whether enasidenib may also show efficacy in IDH-mutant solid tumors.
SDH-deficient GISTs show limited response to first-line imatinib [108] but sunitinib has demonstrated some activity in these patients. Drug screens of >200,000 compounds in SDH-deficient yeast models of paraganglioma identified compounds that were selectively toxic in SDH-mutant but not wild-type yeast [109]. The investigators hypothesized that for a subset of these drugs, inhibition of yeast alcohol dehydrogenase (equivalent to human lactate dehydrogenase), which SDH-deficient cells use to generate NAD+, may be related to the growth inhibitory effect in SDH-deficient yeast and human tumor cell models [109]. Similar to the situation in SDH-deficient GIST, however, clinically effective drugs specifically targeting SDH deficiency in familial paraganglioma/pheochromocytoma remain to be identified.
Novel therapeutic approaches for FH-deficient renal cell carcinoma are subject of ongoing investigation or being tested in clinical trials, and include using metformin to reverse inactivation of AMP-activated protein kinase, inhibition of glucose transport, lactate dehydrogenase A, the antioxidant response pathway, the heme oxygenase pathway, and targeting the tumor vasculature and glucose transport with agents such as bevacizumab and erlotinib [110].
Deeper insights into the metabolic and epigenetic alterations in IDH-, SDH-, and FH-deficient tumors are required in order to identify dependencies and vulnerabilities to develop overarching therapeutic concepts that may be applied to more than one tumor type.
Concluding remarks and perspective
The discovery of mutations in genes encoding the tricarboxylic acid cycle enzymes IDH1/2, SDH, and FH in cancer has expanded our understanding of the complex regulation of metabolism in cancer cells, and provided insight into oncogenic and tumor suppressor properties of mutant metabolic enzymes. Mutant IDH1/2, SDH, and FH share common effects on global DNA methylation and histone modification; further studies are needed to determine the exact mechanisms leading to tumor development and to identify potential targets for treatment.
Biologic context and timing of mutation (for instance, as demonstrated by the model of somatic mosaicism) further play a role in the different clinical presentations of affected individuals. The fact that mutations in the same gene may lead to different phenotypic manifestations (pleiotropy) and that one particular tumor type can be caused by mutations in different genes (genetic heterogeneity) offers the opportunity to better characterize the genetic underpinnings of mesenchymal tumors arising in patient with germline or somatic mosaic mutations in metabolic enzymes.
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Warburg O. On the origin of cancer cells. Science. 1956;123:309–14.
Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–90.
Losman JA, Kaelin WG Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–52.
Pansuriya TC, van Eijk R, d’Adamo P, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–61.
Amary MF, Damato S, Halai D, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011;43:1262–5.
Eng C, Kiuru M, Fernandez MJ, et al. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202.
Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108:314–8.
Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–10.
Krebs HA, Johnson WA. Metabolism of ketonic acids in animal tissues. Biochem J. 1937;31:645–60.
Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem Biophys. 2014;68:475–8.
Laurenti G, Tennant DA. Isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH), fumarate hydratase (FH): three players for one phenotype in cancer? Biochem Soc Trans. 2016;44:1111–6.
Dalziel K, Isocitrate dehydrogenase and related oxidative decarboxylases. FEBS Lett. 1980;117(Suppl):K45–55.
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12.
Paschka P, Schlenk RF, Gaidzik VI, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–43.
Medeiros BC, Fathi AT, DiNardo CD, et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31:272–81.
Jo VY, Chau NG, Hornick JL, et al. Recurrent IDH2 R172X mutations in sinonasal undifferentiated carcinoma. Mod Pathol. 2017;30:650–9.
Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–9.
Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–43.
Kurek KC, Pansuriya TC, van Ruler MA, et al. R132C IDH1 mutations are found in spindle cell hemangiomas and not in other vascular tumors or malformations. Am J Pathol. 2013;182: 1494–1500.
Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009;27:4150–4.
Nobusawa S, Watanabe T, Kleihues P, et al. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res. 2009;15:6002–7.
Hartmann C, Meyer J, Balss J, Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469–74.
Goyal L, Govindan A, Sheth RA, et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist. 2015;20:1019–27.
DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90:732–6.
Cleven AHG, Suijker J, Agrogiannis G, et al. IDH1 or -2 mutations do not predict outcome and do not cause loss of 5-hydroxymethylcytosine or altered histone modifications in central chondrosarcomas. Clin Sarcoma Res. 2017;7:8
Fujii Y, Ogasawara S, Oki H, et al. A high-sensitive HMab-2 specifically detects IDH1-R132H, the most common IDH mutation in gliomas. Biochem Biophys Res Commun. 2015;466: 733–9.
Fletcher CDM. WHO classification of tumours of soft tissue and bone. IARC Press, Lyon, 2013.
Schrage YM, Lam S, Jochemsen AG, et al. Central chondrosarcoma progression is associated with pRb pathway alterations: CDK4 down-regulation and p16 overexpression inhibit cell growth in vitro. J Cell Mol Med. 2009;13:2843–52.
Tarpey PS, Behjati S, Cooke SL, et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet. 2013;45:923–6.
Totoki Y, Yoshida A, Hosoda F, et al. Unique mutation portraits and frequent COL2A1 gene alteration in chondrosarcoma. Genome Res. 2014;24:1411–20.
Zhang YX, van Oosterwijk JG, Sicinska E, et al. Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin Cancer Res. 2013;19:3796–807.
Bovee JV, Hogendoorn PC, Wunder JS, et al. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer. 2010;10:481–8.
Pansuriya TC, Kroon HM, Bovee JV. Enchondromatosis: insights on the different subtypes. Int J Clin Exp Pathol. 2010;3:557–69.
Verdegaal SH, Bovee JV, Pansuriya TC, et al. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: an international multicenter study of 161 patients. Oncologist. 2011;16:1771–9.
Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34.
Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–44.
Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:966.
Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30.
Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Suijker J, Baelde HJ, Roelofs H, et al. The oncometabolite D-2-hydroxyglutarate induced by mutant IDH1 or -2 blocks osteoblast differentiation in vitro and in vivo. Oncotarget. 2015;6:14832–42.
Jin Y, Elalaf H, Watanabe M, et al. Mutant IDH1 dysregulates the differentiation of mesenchymal stem cells in association with gene-specific histone modifications to cartilage- and bone-related henes. PLoS ONE. 2015;10:e0131998.
Jin G, Reitman ZJ, Duncan CG, et al. Disruption of wild-type IDH1 suppresses D-2-hydroxyglutarate production in IDH1-mutated gliomas. Cancer Res. 2013;73:496–501.
Suijker J, Oosting J, Koornneef A, et al. Inhibition of mutant IDH1 decreases D-2-HG levels without affecting tumorigenic properties of chondrosarcoma cell lines. Oncotarget. 2015;6:12505–19.
Li L, Paz AC, Wilky BA, et al. Treatment with a small molecule mutant IDH1 inhibitor suppresses tumorigenic activity and decreases production of the oncometabolite 2-hydroxyglutarate in human chondrosarcoma cells. PLoS ONE. 2015;10:e0133813.
Cleven AH, Zwartkruis E, Hogendoorn PC, et al. Periosteal chondrosarcoma: a histopathological and molecular analysis of a rare chondrosarcoma subtype. Histopathology. 2015;67:483–90.
Damato S, Alorjani M, Bonar F, et al. IDH1 mutations are not found in cartilaginous tumours other than central and periosteal chondrosarcomas and enchondromas. Histopathology. 2012;60: 363–5.
Arai M, Nobusawa S, Ikota H, et al. Frequent IDH1/2 mutations in intracranial chondrosarcoma: a possible diagnostic clue for its differentiation from chordoma. Brain Tumor Pathol. 2012;29:201–6.
Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88.
Stratakis CA, Carney JA. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. J Intern Med. 2009;266:43–52.
Matyakhina L, Bei TA, McWhinney SR, et al. Genetics of carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J Clin Endocrinol Metab. 2007;92: 2938–43.
Chase WH. Familial and bilateral tumours of the carotid body. J Pathol Bacteriol. 1933;36:1–12.
Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–33.
Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–66.
Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med. 2009;266:19–42.
Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–20.
Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–33.
Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–7.
Fishbein L, Leshchiner I, Walter V, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31:181–93.
Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54.
Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–70.
Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–51.
Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–42.
Benn DE, Robinson BG, Clifton-Bligh RJ. 15 years of paraganglioma: clinical manifestations of paraganglioma syndromes types 1-5. Endocr Relat Cancer. 2015;22:T91–103.
Castro-Vega LJ, Buffet A, De Cubas AA, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–6.
Clark GR, Sciacovelli M, Gaude E, et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab. 2014;99:E2046–50.
Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–52.
Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277–85.
Gupta S, Zhang J, Milosevic D, et al. Primary renal paragangliomas and renal neoplasia associated with pheochromocytoma/paraganglioma: analysis of von Hippel-Lindau (VHL), succinate dehydrogenase (SDHX) and transmembrane protein 127 (TMEM127). Endocr Pathol. 2017;28:253–268.
Shuch B, Linehan WM, Srinivasan R. Aerobic glycolysis: a novel target in kidney cancer. Expert Rev Anticancer Ther. 2013;13:711–719.
Ricketts CJ, Shuch B, Vocke CD, et al. Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol. 2012;188:2063–71.
Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol. 2015;28:80–94.
Smith SC, Sirohi D, Ohe C, et al. A distinctive, low-grade oncocytic fumarate hydratase-deficient renal cell carcinoma, morphologically reminiscent of succinate dehydrogenase-deficient renal cell carcinoma. Histopathology. 2017;71:42–52.
Miettinen M, Killian JK, Wang ZF, et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol. 2013;37:234–40.
Wagner AJ, Remillard SP, Zhang YX, et al. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod Pathol. 2013;26:289–94.
Oudijk L, Gaal J, Korpershoek E, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013;26:456–63.
Xekouki P, Stratakis CA. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer. 2012;19:C33–40.
Jiang Q, Zhang Y, Zhou YH, et al. A novel germline mutation in SDHA identified in a rare case of gastrointestinal stromal tumor complicated with renal cell carcinoma. Int J Clin Exp Pathol. 2015;8:12188–97.
Doyle LA, Nelson D, Heinrich MC, et al. Loss of succinate dehydrogenase subunit B (SDHB) expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumours: a comprehensive genotype-phenotype correlation study. Histopathology. 2012;61:801–9.
Haller F, Moskalev EA, Faucz FR, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer. 2014;21:567–77.
Killian JK, Miettinen M, Walker RL, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6:268ra177
Boikos SA, Xekouki P, Fumagalli E, et al. Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur J Hum Genet. 2016;24:569–73.
Mason EF, Hornick JL. Conventional risk stratification fails to predict progression of succinate dehydrogenase-deficient gastrointestinal stromal tumors: a clinicopathologic study of 76 cases. Am J Surg Pathol. 2016;40:1616–21.
Smith EH, Janknecht R, Maher LJ III. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16:3136–48.
Xiao M, Yang H, Xu W, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–38.
Cervera AM, Bayley JP, Devilee P, et al. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol Cancer. 2009;8:89.
Boikos SA, Pappo AS, Killian JK, et al. Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors: a report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2:922–8.
Mason EF, Hornick JL. Succinate dehydrogenase deficiency is associated with decreased 5-hydroxymethylcytosine production in gastrointestinal stromal tumors: implications for mechanisms of tumorigenesis. Mod Pathol. 2013;26:1492–7.
Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget. 2015;6:38777–88.
Pithukpakorn M, Toro JR. Hereditary leiomyomatosis and renal cell cancer. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. Gene Reviews(R). Seattle, WA, University of Washington, 1993.
Miettinen M, Felisiak-Golabek A, Wasag B, et al. Fumarase-deficient uterine Leiomyomas: an immunohistochemical, molecular genetic, and clinicopathologic study of 86 cases. Am J Surg Pathol. 2016;40:1661–9.
Joseph NM, Solomon DA, Frizzell N, et al. Morphology and immunohistochemistry for 2SC and FH aid in detection of fumarate hydratase gene aberrations in uterine Leiomyomas from young patients. Am J Surg Pathol. 2015;39:1529–39.
Reyes C, Karamurzin Y, Frizzell N, et al. Uterine smooth muscle tumors with features suggesting fumarate hydratase aberration: detailed morphologic analysis and correlation with S-(2-succino)-cysteine immunohistochemistry. Mod Pathol. 2014;27:1020–7.
Bardella C, El-Bahrawy M, Frizzell N, et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011;225:4–11.
Ternette N, Yang M, Laroyia M, et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013;3:689–700.
Llamas-Velasco M, Requena L, Adam J, et al. Loss of fumarate hydratase and aberrant protein succination detected with S-(2-succino)-cysteine staining to identify patients with multiple cutaneous and uterine Leiomyomatosis and hereditary Leiomyomatosis and renal cell cancer syndrome. Am J Dermatopathol. 2016;38:887–91.
Killian JK, Kim SY, Miettinen M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3:648–57.
Wentzel JF, Lewies A, Bronkhorst AJ, et al. Exposure to high levels of fumarate and succinate leads to apoptotic cytotoxicity and altered global DNA methylation profiles in vitro. Biochimie. 2017;135:28–34.
Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–8.
McDonough MA, Loenarz C, Chowdhury R, et al. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659–72.
Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–9.
Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–53.
Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85.
Popovici-Muller J, Saunders JO, Salituro FG, et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med Chem Lett. 2012;3:850–5.
Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–30.
Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–6.
Kim ES. Enasidenib: first global approval. Drugs. 2017;77:1705–1711.
Miettinen M, Wang ZF, Sarlomo-Rikala M, et al. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35:1712–21.
Bancos I, Bida JP, Tian D, et al. High-throughput screening for growth inhibitors using a yeast model of familial paraganglioma. PLoS ONE. 2013;8:e56827.
Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase-deficient kidney cancer--targeting the Warburg effect in cancer. Clin Cancer Res. 2013;19:3345–52.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Confict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Schaefer, IM., Hornick, J.L. & Bovée, J.V.M.G. The role of metabolic enzymes in mesenchymal tumors and tumor syndromes: genetics, pathology, and molecular mechanisms. Lab Invest 98, 414–426 (2018). https://doi.org/10.1038/s41374-017-0003-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-017-0003-6
This article is cited by
-
Maffucci syndrome complicated by giant chondrosarcoma in the left ankle with an IDH1 R132C mutation: a case report
World Journal of Surgical Oncology (2022)
-
Atypical cartilage in type II germ cell tumors of the mediastinum show significantly different patterns of IDH1/2 mutations from conventional chondrosarcoma
Modern Pathology (2022)
-
What Have We Learned from Molecular Biology of Paragangliomas and Pheochromocytomas?
Endocrine Pathology (2021)
-
The evolving metabolic landscape of chromatin biology and epigenetics
Nature Reviews Genetics (2020)
-
Development of somatic mutation signatures for risk stratification and prognosis in lung and colorectal adenocarcinomas
BMC Medical Genomics (2019)
