
Abstract
Cancer epigenetics is one of the most important research subjects in dissecting cancer mechanisms and therapeutic targets because the emergence and malignant transformation of various cancers are caused by unnatural expression of cancer-related genes attributed to their epigenetic errors. The original concept of cancer epigenetics basically stands on the analysis of the epigenetic status in naturally occurring cancer cells; however, the rapidly emerging technology called epigenome editing would change this situation drastically. Epigenome editing, the most promising derivative technology of genome editing, can modify the epigenetic states at the pre-defined genomic locus using the programmable effectors, consisting of various epigenetic factors combined with site-specific DNA-binding domains. This technology can be utilized in a reversible manner; i.e., cancer modeling can be achieved by introducing aberrant epigenetic marks in normal cells, and cancer suppression can be achieved by correcting the epigenetic errors in cancer cells. In this review, we summarize the basics of epigenome editing and cancer epigenetics, followed by the current examples of cancer induction and suppression with the transcriptional control and epigenome editing technologies.
Similar content being viewed by others

Introduction
Recent definition of epigenetics can be expressed as the mechanisms of regulating gene expression along with the genomic DNA sequence [1] such as DNA methylation [2, 3], histone modification [3, 4], non-coding RNA [5], and chromatin higher-order structures [6]. These features regulate global or local gene expression via various modes of action, and mostly inherit after cell division. Epigenetics studies are essential to elucidate diverse life phenomena; thus, a huge variety of analytical techniques has been developed. For example, bisulfite sequencing [7] detects individual methylated cytosine in the target genomic region, and chromatin immunoprecipitation (ChIP) assay [8,9,10] detects DNA sequences interacting with specific proteins such as modified histone. Furthermore, recently developed techniques such as ChIP-seq [11], Infinium [12, 13], and 3C-based methods [14] have allowed global analysis of various epigenetic states.
Epigenetic errors cause various human diseases such as cancer, protein aggregation diseases, metabolic diseases, neurological and psychiatric diseases, and imprinting disorders [15]. As can be expected from the past studies, it has become available to analyze such natural epigenetic errors found in epigenetic diseases, including cancers, which can be expressed as “forward epigenetics”. Yet, “reverse epigenetics”, i.e., analysis of local epigenetic functions by artificially modifying the target genomic region was extremely difficult because the appropriate research tools for such analysis had been unavailable. Epigenome editing is the one and only technology enabling reverse epigenetics; thus, it will open a new avenue for cancer epigenetics study. In this review, we summarize the current transcriptional control and epigenome editing technology with focusing on the cancer induction and suppression.
Cancer epigenetics
Epigenetics study, especially for the chemical tags of DNA and histone, has historically been closely related to cancer research. Cancer epigenetics was first discovered as the significant reduction of DNA methylation in human tumors [16, 17]. Current understanding is that the genomic DNA in cancer cells is globally demethylated, whereas abnormally methylated DNA can be found at the CpG islands (CGIs) in the regulatory region of tumor suppressor genes (TSGs), causing their impaired expression. It also has proved by numerous studies that abnormal histone modifications around cancer-related genes affect cancer induction and progression. In addition, mutations of epigenetic modifier genes such as DNMT (DNA methyltransferase), KAT (histone acetyltransferase), and KMT (histone methyltransferase) have been known to cause global epigenetic changes, which have been observed in a wide variety of cancers [18]. These basic findings of cancer epigenetics have been well summarized in magnificent previous reviews [18,19,20,21].
Non-coding RNA and chromatin higher-order structure are also key mechanisms to understand cancer induction and progression. Non-coding RNA, which is a collective term of the RNA molecules not encoding a protein, has various biological functions, including transcription, RNA splicing, and translation [5, 22]. The microRNA (miRNA) and T-UCR (transcribed ultraconserved regions), both categorized as non-coding RNA, upregulate the cancer metastatic ability and induce epithelial-mesenchymal transition (EMT). Long non-coding RNA (lncRNA) promotes reconstruction of chromatin structure and progresses cancer via transcriptional regulation. In addition, chromatin higher-order structure such as topologically associated domains (TAD) also regulates gene expression [6]. Regarding the relationship between TAD and cancer, for example, TAD-associated CTCF sites are known to be highly mutated compared to other CTCF sites in esophageal and liver carcinoma [23].
The frameworks of epigenome editing tools
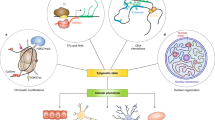
The current epigenome editing technology has been focused on modifying the chemical tags of DNA and histones. The basic architecture of epigenome editing tools depends on a chimeric protein consisting of epigenetic effector domain and programmable DNA-binding domain such as such as zinc finger (ZF) [24] (Fig. 1a), transcription activator-like effector (TALE) [25] (Fig. 1b), and nuclease-deficient Cas9 (dCas9) [26, 27] (Fig. 1c). Thus, epigenome editing enzymes can be regarded as the variant of genome editing nucleases, replacing the nuclease activity with the epigenetic effector activity.

Programmable systems for transcriptional control and epigenome editing. The first-generation systems depend on the direct linkage of the epigenetic effector and the DNA-binding proteins such as ZF (a), TALE (b), and dCas9 (c). In the second-generation systems, synergistic activation can be achieved by recruiting multiple effectors via dCas9-SunTag d and SAM e systems
Classical genome editing tools such as ZF nuclease (ZFN) [28] and TALE nuclease (TALEN) [29] are composed of the protein-based DNA-binding motifs and the nuclease domain derived from FokI enzyme. Natural ZFs can be found in various transcription factors and regulatory proteins in a wide variety of eukaryotic organisms, whose conformation is surprisingly compact and sophisticated. ZFs have been used as a platform for epigenome editing in the early days [30,31,32,33]; however, artificial engineering of ZFs is very difficult because the base-recognition specificity of ZFs can change in a context-dependent manner, when the motifs are tandemly assembled. The TALE protein, the DNA-binding domain of the second-generation genome editing tool, has also utilized for epigenome editing [34,35,36]. TALEs have the DNA-binding repeats that can recognize DNA bases on a one-to-one basis, whose base-recognition specificity is rarely affected by the context. Therefore, TALEs are much more easily customizable than ZFs. Since ZF- or TALE-based epigenome editing reagents consist of protein alone, they might be simply delivered for clinical applications as the protein drugs.
Clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated protein 9 (Cas9) [37, 38], the easiest-to-use nuclease for genome editing, works as a complex of a chimeric single guide RNA (sgRNA) and Cas9 nuclease. The sgRNA recognizes around 20-base DNA sequence, which is easily and freely programmable. Cas9 recognize a few bases called protospacer adjacent motif (PAM), which is barely programmable. The PAM sequence of Cas9 derived from Streptococcus pyogenes (SpCas9), which is the most commonly used Cas9, is 5′-NGG-3′. The specificity of the PAM sequence is different among species, and a few examples of artificial alteration of PAM specificity have been reported [39]. Thus, any sequence can be targeted by CRISPR-Cas9 (i.e., gene-specific sgRNA and common Cas9 protein) except for the PAM restriction. The wild-type Cas9 protein harbors the nuclease activity, which is unnecessary for epigenome editing. Therefore, epigenetic effectors are fused with catalytically-dead dCas9. Collectively, CRISPR-based epigenome editing can be performed with the gene-specific CRISPR-dCas9 reagent, consisting of synthesis-friendly sgRNA and generic Cas9; thus, the creation of the materials is rather simple and cost-effective compared with the ZF- and TALE-based tools. This feature is practically quite advantageous, and the technical development of epigenome editing has rapidly been evolving with CRISPR-dCas9 platform [40,41,42,43,44].
Transcriptional control with artificial transcription factors
For the transient regulation of transcription, artificial transcription factors (ATFs), transcriptional activator or repressor fused with programmable DNA-binding domain, have been used instead of direct alteration of epigenetics [45,46,47]. In principle, such ATFs can commonly work in the promoter region of the gene of interest, while epigenetic effectors should be designed corresponding to the positions of endogenous epigenetic marks, which vary with the variety of every gene. In this regard, ATFs are technically easy to apply; thus, they are developed as underlying technology of epigenome editing.
Synthetic ATFs can be divided into two types according to their functions; transcriptional activator and suppressor. Typical activation domains are VP16 derived from herpes simplex virus [45] and its concatemer (VP64 [46] and VP160 [48]). For the suppressors, Krüppel-associated box (KRAB) domain [45] and mSin3 interaction domain (SID) [49] are often used. Of these, transcriptional suppression mediated by KRAB domain has frequently been reported in cancer modeling and therapeutic approaches, including the induction of breast cancer [31] and the suppression of several cancers including ovarian [33], lung [50], and monocytic leukemia [51].
However, the primary design of ATFs only brings one effector domain per one DNA-binding molecule. Therefore, more effective second-generation systems have been invented by several groups such as dCas9-SunTag [52] and synergistic activation mediators (SAM) [53] (Fig. 1d) systems. These systems can accumulate multiple activators at the dCas9-binding sites. In addition, strong activation was also achieved by the artificial fusion activator, VP64-p65-Rta (VPR) [54]. Particularly, as explained below, SAM system was applied to construct the potential therapeutic model in cancer cells [55]. SAM system consists of three components; dCas9 fused with VP64 (dCas9-VP64), sgRNA containing two MS2 RNA aptamers (sgRNAMS2), and chimeric activator fused with MS2 coat protein (MS2-p65-HSF1). MS2-p65-HSF1 is captured at the MS2 loops of sgRNAMS2; thus, VP64 and p65-HSF1 can act as synergistic activators at the dCas9-binding regions.
Expanded applications using the synthetic ATF systems
Synthetic ATFs have been applied to cancer induction and suppression in various ways. Blancafort and colleagues showed that SAM system could increase the REPRIMO mRNA expression (680-fold change) in AGS cell line derived from stomach cancer [55]. REPRIMO is a tumor suppressor gene that triggers G2 arrest in a tp53-dependent manner; thus, the activation of REPRIMO decreases cell viability. In parallel with the above mechanisms, the decreased and increased expression of proliferation marker Ki67 and apoptotic marker Caspase-3, respectively, were also observed in the REPRIMO-activated cells. These observations indicate that the activation of REPRIMO gene induces tumor suppression in vitro. Moreover, using the modified version of SAM system, containing VPR fused with dCas9 (dCas9-VPR) instead of dCas9-VP64, 22,145-fold activation of MASPIN mRNA expression in H157 lung cancer cells was achieved, which was more effective than the treatment of dCas9-VPR without the synergistic activator.
Huang and Cai groups showed that the expression of tumor suppressor p53 gene could be upregulated by light-inducible ATFs with exposure to blue light in 5637 and UMUC-3 bladder cancer cells, resulting in the suppression of cell proliferation [56]. According to their validation study, dCas9 fused with two truncated CIB1 (CIBN) modules (CIBN-dCas9-CIBN) in combination with photolyase homology region of CRY2 (CRY2PHR) fused with p65 activation domain (CRY2PHR-p65) activated p53 mRNA expression most efficiently, where 20–30% reduction of cell proliferation was observed.
In vivo cancer induction using ATFs was demonstrated by Hemann and colleagues, by regulating the transcription of cancer-related genes such as Trp53 and Mgmt using dCas9 and dCas9-VP64 [57]. Trp53 gene was silenced by dCas9/sgRNA designed to target downstream of transcription start site in lymphoma cells, and then the cells were transplanted into recipient mice. In the presence of dCas9/sgRNA, time to disease onset became shorter than control mice. Overall, survival period also became shorter with or without cisplatin administration. Similarly, Mgmt upregulation by dCas9-VP64/sgRNA in leukemia cells followed by transplantation resulted in reduced survival rate with temozolomide; however, it did not show any difference in survival rate in the absence of treatment. Thus, they showed in vivo modeling of cancer progression and therapeutic relapse by transcriptional control systems.
Cancer induction and suppression with bona fide epigenome editing
In the bona fide epigenome editing, i.e., removing, adding, and modifying epigenetic marks such as DNA and histone tags, similar systems have been adopted. Most studies have utilized simple linking of DNA-binding module and epigenetic effector, but some groups recently reported the application of the second-generation architecture, SAM [58] and SunTag [59], in epigenome editing.
From here we selectively introduce various milestone studies of cancer induction and suppression, mediated by epigenome editing technologies. The selective list of such studies is also summarized in Table 1; however, note that the following text is written in the context of effector domains, while the list is categorized by cancer induction/suppression and cancer types in Table 1. For the comprehensive list of epigenome editing applications categorized by effector domains, see the previous review by Beck and colleagues [60].
Targeted DNA methylation and demethylation
The examinations of epigenome editing with DNA methylation/demethylation have been reported by many groups, where TET1 demethylase [36, 41, 58, 59, 61] and DNMT3A methyltransferase [31, 32, 41, 43, 44,62,63,64] have been used as the epigenetic effectors.
TET1 DNA demethylase has been used for epigenome editing from a relatively early stage of this field of study [36, 41, 61]. Human TET1 is a large protein exceeding 2000 amino acids (a.a.), while the study of targeted DNA demethylation based on the TALE technology revealed that 700-a.a. catalytic domain of TET1 (TET1CD) could efficiently induce DNA demethylation [36]. After that, other epigenetic effectors have been often used compactly by removing extra domains other than the catalytic domain. Stefanska, Irudayaraj, and colleagues reported a therapeutic application of dCas9-TET1CD [61], in which they induced the increased expression of a TSG, BRCA1 (up to about 2.5-fold change), in HeLa cervical cancer cells. They also observed 10–15% decrease in the methylation level of the CpG sites at the BRCA1 promoter region.
Several independent studies reported the therapeutic models of cancers, harnessing epigenome editing with DNMT3A. DNMT3A is a DNA methyltransferase working in de novo methylation. DNMT3A-mediated tumor suppression was shown by introducing DNA methylation at the promoter regions of oncogenes in MCF7 cells [64], SKOV3 ovarian cancer cells [32], and A549 lung cancer cells [50]. One especially notable research is SOX2 silencing by Blancafort and colleagues [64], showing the tumor growth suppression in vivo by methylating DNA using ZF-DNMT3A (fusion protein of ZF and the catalytic domain of DNMT3A) targeting the SOX2 promoter. They first established the MCF7 cells with the doxycycline-inducible expression of ZF-DNMT3A, and then implanted it into a nude mouse. After the sufficient growth of the tumor, doxycycline was administered into the xenograft mouse to express ZF-DNMT3A, aiming for the reduction of tumor progression by increasing DNA methylation at the SOX2 promoter. Indeed, tumor phenotype was dramatically changed with ZF-DNMT3A, with the decrease and increase of mesenchymal markers (SOX2, TWIST1, and Vimentin) and epithelial junction protein (Claudin 4), respectively. Furthermore, after doxycycline treatment, the tumors formed more organized clusters and the extended tumor gaps were observed histologically. This study is the excellent example of site-specific in vivo DNA methylation to possibly contribute cancer therapy.
Moreover, recent studies showed that the combinatorial recruitment of DNMT3A and DNMT3L is more effective than DNMT3A alone [32, 44, 62]. DNMT3L does not have catalytic activity, but it reportedly stimulates de novo methyltransferases (i.e., DNMT3A and DNMT3B). Cancer modeling using DNMT3L has been reported by Kaestner and colleagues, showing that CDKN2A downregulation by methylating its promoter region upregulated cell proliferation of fibroblast cells [62]. Consistently, high-efficiency and long-term DNA methylation were induced using dCas9 or TALE combined with DNMT3L, DNMT3A, and KRAB domain, as shown by Lombardo and colleagues [44]. They showed silencing of B2M gene by the triple effectors in K562 and HEK293T cells for more than 50 days. The stability of epigenome editing outcome is an important factor especially for the cancer therapy, because repeated administration of epigenetic effectors significantly increase the medical expenses and physical and emotional strains. Accordingly, we have to consider how long the epigenetic modification is maintained with every effector, at every target locus, and in every cell type, for the purpose of therapeutic use.
Targeted histone modifications
Since there are a wide variety of histone modifications mediated by numerous effectors, programmable enzymes targeting histone tags are also variable compared to those for the DNA modification; e.g., histone demethylase (LSD1 [34, 42]), histone methyltransferases (PRDM9 [40], G9a [30, 33, 50, 65] and SUV39H1 [30, 33]), and histone acetyltransferase (p300) [66].
Especially, G9a, also known as EHMT2, is a lysine 9 of histone H3 methyltransferase, which has been proven to be utilized in cancer modeling [65] and suppression [33, 50] as a programmable enzyme. Kim and colleagues demonstrated the cancer modeling with the catalytic SET domain of G9a [65], by showing that cancer malignancy was enhanced in HeLa and HCT116 colon cancer cells. Since E-cadherin decreases when cancer cells make the shift to epithelial-mesenchymal transition (EMT), they suppressed the expression of CDH1 gene encoding E-cadherin protein using TALE-SET (TSET). The silencing of CDH1 upregulated the migration and invasion activities of HeLa and HCT116 cells. Additionally, they observed increased H3K9me2 in CDH1 promoter, whereas the H3K4me2 level was not changed in HeLa cells.
Cancer suppression with G9a was reported by Rots and Hylkema groups [50]. They introduced de novo histone methylation using dCas9-G9a (catalytic domain of G9a fused with dCas9) in A549 lung cancer cells. dCas9-G9a was designed to target the promoter region of SPDEF gene, involved in mucus production in lung epithelial cells. SPDEF suppression also induced decreased expression of other mucus producer genes such as AGR2 and MUC5AC. They also indicated that H3K9 methylation using dCas9-G9a could enable long-term suppression of SPDEF expression compared to direct transcriptional repression (dCas9-KRAB) and DNA methylation (dCas9-DNMT3A-DNMT3L).
The Suvdel76 histone methylation domain, the N-terminally deleted protein of SUV39H1, was also used for the potentially therapeutic purpose [33]. Rots and colleagues showed that HER2/neu genes, which encode EGF receptor family and regulate cell proliferation pathway, were silenced by ZF-Suvdel76 and ZF-G9a in breast cancer cells (MCF7 and SKBR3) and ovarian cancer cells (SKOV3), resulting in reduced cell proliferation and metabolic activities.
The core domain of p300 (p300cd), one of the histone acetyltransferases, has not yet been utilized in cancer induction or suppression; however, it is also a critically important effector in epigenome editing. The activation of the target gene was achieved by directing proximal and distal enhancer with dCas9-p300cd, which could not be realized using dCas9-VP64 [66]. On the basis of this, Cas9-based epigenomic regulatory element screening (CERES) was also reported [67]. The CERES method enabled both gain- and loss-of-function screenings with the following procedures. First, the gene of interest (GOI) was tagged with a fluorescent protein gene to monitor endogenous gene expression. Subsequently, the cells were infected with lentivirus carrying dCas9-KRAB or dCas9-p300cd. After establishing the stable cell clones, lentiviral sgRNA library was infected into these cells. The library was designed to target potential regulatory elements of the GOI, predicted by DNase I hypersensitive sites. Finally, the cells with changes in the expression of the GOI were selected using fluorescence-activated cell sorting (FACS), and the population of sgRNAs was analyzed by next-generation sequencing to determine the functional regulatory elements.
The CERES analysis revealed the transcriptional regulatory elements of HER2 gene, which is an adverse prognostic factor in breast cancer. The results indicated that the active regulatory elements were located mostly around the promoter, but previously unknown elements were also identified. Furthermore, it was found that a part of regulatory elements identified with CERES analysis were different in accordance with the cell types and effector domains.
Conclusions
Epigenome editing technology has enabled “reverse epigenetics” analysis, which clarifies region-specific function of cancer epigenetics. Although the current epigenome editing studies generally target promoter regions, the target sequence suitable for epigenetic modifications, followed by the activation or repression of the gene of interest, is not limited in typical promoters, as CERES study suggested. Given that we set the cancer therapy one of the goals of epigenome editing technology, exploring the regulatory elements in each cancer type or each target gene will fundamentally be required, as well as the technical development of editing systems.
For the clinical application of this technology, delivery challenge is another important and critical issue to realize in vivo cancer therapy. Adeno-associated virus (AAV)-mediated delivery has been considered as a mainstream technique. For the selective transduction in cancers, various improved AAVs have been reported by targeting tumor-specific antigens [68]. Additionally, non-viral delivery mediated by lipid nanoparticles is also a promising technique [69]. Nevertheless, epigenome editing tools should ideally be introduced into all the cancer cells; thus, there is still a hurdle to translate this technology into clinics.
Currently, epigenome editing is virtually a synonym for targeted modification of chemical tags; however, programmable control of non-coding RNA and chromatin organization should also be developed and included in epigenome editing techniques. The non-coding RNAs might be able to regulate by utilizing the RNA-guided RNA endonucleases such as Cas13a [70]. Chromatin higher-order structures include biologically complex mechanisms, but recent study showed that chromosomal looping can be triggered with the CRISPR tool [71]. These new tools and methods will also open new doors for cancer epigenetics and potential therapy.
References
Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500.
Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76.
Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390–403.
Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–31.
Kuo MH, Allis CD. In vivo cross-linking and immunoprecipitation for studying dynamic Protein: DNA associations in a chromatin environment. Methods. 1999;19:425–33.
Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–14.
Thorne AW, Myers FA, Hebbes TR. Native chromatin immunoprecipitation. Methods Mol Biol. 2004;287:21–44.
Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–80.
Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8:389–99.
Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina Methylation EPIC Bead Chip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208.
Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11:476–86.
Kungulovski G, Jeltsch A. Epigenome editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016;32:101–13.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92.
Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, et al. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–94.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53.
Baylin SB, Ohm JE. Epigenetic gene silencing in cancer–a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–16.
Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98.
Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15:R17–29.
Hnisz D, Weintraub A, Day DS, Valton AL, Bak RO, Li C, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351:1454–8.
Berg JM. Proposed structure for the zinc-binding domains from transcription factor IIIA and related proteins. Proc Natl Acad Sci. 1988;85:99–102.
Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: finding plant genes for disease and defense. Curr Opin Plant Biol. 2010;13:394–401.
Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41:7429–37.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83.
Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci. 1996;93:1156–60.
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–61.
Snowden AW, Gregory PD, Case CC, Pabo CO. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr Biol. 2002;12:2159–66.
Rivenbark AG, Stolzenburg S, Beltran AS, Yuan X, Rots MG, Strahl BD, et al. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics. 2012;7:350–60.
Siddique AN, Nunna S, Rajavelu A, Zhang Y, Jurkowska RZ, Reinhardt R, et al. Targeted methylation and gene silencing of VEGF‑A in human cells by using a designed Dnmt3a‑Dnmt3L single-chain fusion protein with increased DNA methylation activity. J Mol Biol. 2013;425:479–91.
Falahi F, Huisman C, Kazemier HG, van der Vlies P, Kok K, Hospers GA, et al. Towards sustained silencing of HER2/neu in cancer by epigenetic editing. Mol Cancer Res. 2013;11:1029–39.
Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, et al. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. 2013;31:1133–6.
Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–6.
Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31:1137–42.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
Nakade S, Yamamoto T, Sakuma T. Cas9, Cpf1 and C2c1/2/3-What’s next? Bioengineered. 2017;4:265–73.
Cano-Rodriguez D, Gjaltema RA, Jilderda LJ, Jellema P, Dokter-Fokkens J, Ruiters MH, et al. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat Commun. 2016;7:12284.
Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–47.
Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, et al. Functional annotation of native enhancers with a Cas9‑histone demethylase fusion. Nat Methods. 2015;12:401–3.
Vojta A, Dobrinić P, Tadić V, Bočkor L, Korać P, Julg B, et al. Repurposing the CRISPR–Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–28.
Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, et al. Inheritable silencing of endogenous genes by hit-and-run targeted epigenetic editing. Cell. 2016;167:219–32.
Liu Q, Segal DJ, Ghiara JB, Barbas CF. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc Natl Acad Sci USA. 1997;94:5525–30.
Beerli RR, Dreier B, Barbas CF. Positive and negative regulation of endogenous genes by designed transcription factors. Proc Natl Acad Sci USA. 2000;97:1495–500.
Davis D, Stokoe D. Zinc finger nucleases as tools to understand and treat human diseases. BMC Med. 2010;8:42.
Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–71.
Beerli RR, Segal DJ, Dreier B, Barbas CF. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci USA. 1998;95:14628–33.
Song J, Cano-Rodriquez D, Winkle M, Gjaltema RA, Goubert D, Jurkowski TP, et al. Targeted epigenetic editing of SPDEF reduces mucus production in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2017;312:L334–47.
Radzisheuskaya A, Shlyueva D, Müller I, Helin K. Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res. 2016;44:e141.
Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–46.
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–8.
Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12:326–8.
Garcia-Bloj B, Moses C, Sgro A, Plani-Lam J, Arooj M, Duffy C, et al. Waking up dormant tumor suppressor genes with zinc fingers, TALEs and the CRISPR/dCas9 system. Oncotarget. 2016;7:60535.
Lin F, Dong L, Wang W, Liu Y, Huang W, Cai Z. An Efficient light-inducible P53 expression system for inhibiting proliferation of bladder cancer cell. Int J Biol Sci. 2016;12:1273–8.
Braun CJ, Bruno PM, Horlbeck MA, Gilbert LA, Weissman JS, Hemann MT. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc Natl Acad Sci USA. 2016;5:E3892–900.
Xu X, Tao Y, Gao X, Zhang L, Li X, Zou W, et al. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016;2:16009.
Morita S, Noguchi H, Horii T, Nakabayashi K, Kimura M, Okamura K, et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat Biotechnol. 2016;34:1060–5.
Stricker SH, Köferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2016;18:51–66.
Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. CRISPR–dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 2016;7:46545.
Bernstein DL, Le Lay JE, Ruano EG, Kaestner KH. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J Clin Invest. 2015;125:1998–2006.
Nunna S, Reinhardt R, Ragozin S, Jeltsch A. Targeted methylation of the epithelial cell adhesion molecule (EpCAM) promoter to silence its expression in ovarian cancer cells. PLoS ONE. 2014;9:e87703.
Stolzenburg S, Beltran AS, Swift-Scanlan T, Rivenbark AG, Rashwan R, Blancafort P. Stable oncogenic silencing in vivo by programmable and targeted de novo DNA methylation in breast cancer. Oncogene. 2015;34:5427–35.
Cho HS, Kang JG, Lee JH, Lee JJ, Jeon SK, Ko JH, et al. Direct regulation of E-cadherin by targeted histone methylation of TALE-SET fusion protein in cancer cells. Oncotarget. 2015;6:23837.
Hilton IB, D’ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–7.
Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB, et al. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol. 2017;35:561–8.
Santiago-Ortiz JL, Schaffer DV. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Control Release. 2016;240:287–301.
Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine. 2016;12:143–61.
Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573.
Morgan SL, Mariano NC, Bermudez A, Arruda NL, Wu F, Luo Y, et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat Commun. 2017;8:15993.
Acknowledgements
The authors were supported by the Practical Research for Innovative Cancer Control from Japan Agency for Medical Research and Development, AMED (to T.S. and T.Y.) and grants from the Japan Society for the Promotion of Science (16J03164 to S.N., 16K18478 to T.S., and 17H01409 to T.Y. and T.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nakade, S., Yamamoto, T. & Sakuma, T. Cancer induction and suppression with transcriptional control and epigenome editing technologies. J Hum Genet 63, 187–194 (2018). https://doi.org/10.1038/s10038-017-0377-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0377-8
This article is cited by
-
Getting personal with epigenetics: towards individual-specific epigenomic imputation with machine learning
Nature Communications (2023)
