
Abstract
Hermansky-Pudlak syndrome (HPS) is an autosomal recessive disease consisting of oculocutaneous albinism and a storage pool deficiency resulting from absent platelet dense bodies. The disorder is genetically heterogeneous. The majority of patients, including members of a large genetic isolate in northwest Puerto Rico, have mutations in HPS1. Another gene, ADTB3A, was shown to cause HPS-2 in two brothers having compound heterozygous mutations that allowed for residual production of the gene product, the β3A subunit of adaptor complex-3 (AP-3). This heterotetrameric complex serves as a coat protein–mediating formation of intracellular vesicles, e.g. the melanosome and platelet dense body, from membranes of the trans-Golgi network. We determined the genomic organization of the human ADTB3A gene, with intron/exon boundaries, and describe a third patient with β3A deficiency. This 5-y-old boy has two nonsense mutations, C1578T (R→X) and G2028T (E→X), which produce no ADTB3A mRNA and no β3A protein. The associated μ3 subunit of AP-3 is also entirely absent. In fibroblasts, the cell biologic concomitant of this deficiency is robust and aberrant trafficking through the plasma membrane of LAMP-3, an integral lysosomal membrane protein normally carried directly to the lysosome. The clinical concomitant is a severe, G-CSF–responsive neutropenia in addition to oculocutaneous albinism and platelet storage pool deficiency. Our findings expand the molecular, cellular, and clinical spectrum of HPS-2 and call for an increased index of suspicion for this diagnosis among patients with features of albinism, bleeding, and neutropenia.
Similar content being viewed by others


Main
HPS consists of oculocutaneous albinism and a bleeding disorder resulting from absence of platelet dense bodies (1–5), which normally release serotonin, calcium, and ADP to trigger a secondary aggregation response (6). Some HPS patients also accumulate intracellular ceroid lipofuscin, a lipid-protein complex, and some suffer from granulomatous colitis or fatal pulmonary fibrosis (2–5). Although HPS exhibits considerable locus heterogeneity (7, 8), only two causative genes have been identified. One gene, HPS1, encodes a 700-amino acid protein whose function remains unknown but is thought to involve formation of new intracellular vesicles such as melanosomes and platelet dense bodies (9–12). HPS patients from northwest Puerto Rico represent the majority of individuals recognized to have the disease (13) and are homozygous for a 16-bp duplication in HPS1(9). In addition, more than 25% of non-Puerto Rican patients have various other mutations in this gene (4, 8). An inbred mouse called pale ear, one of 15 different murine models of HPS with pigment dilution and platelet storage pool deficiency (14, 15), has mutations in the murine homolog of HPS1(16–18).
Mutations in a second gene, ADTB3A, were recently found to result in a subtype of HPS called HPS-2 (19, 20). ADTB3A codes for the β3A subunit of AP-3 (21). This heterotetrameric complex, comprised of β3A, μ3, ς3, and δ3 subunits, serves as a coat protein that concentrates in a donor membrane, binds clathrin, and recruits cargo proteins to become part of the newly formed vesicle (22–26). Those vesicles are thought to include melanosomes in melanocytes and dense bodies in platelets. Two brothers with compound heterozygous mutations in ADTB3A have HPS, i.e. HPS-2 (20). In addition, the pearl mouse has mutations in Ap3b1, the murine homolog of ADTB3A(27).
We now report a third patient with HPS-2, a 5-y-old boy with compound heterozygous nonsense mutations in ADTB3A. The greater severity of this patient's clinical, molecular, and cell biologic findings expands the disease phenotype, and his cultured fibroblasts provide a superior model system in which to study new vesicle formation. We also report the genomic organization of ADTB3A, with intron/exon boundaries.
METHODS
Patients and cell culture.
All patients were enrolled in protocols approved by the National Institute of Child Health and Human Development (NICHD) Institutional Review Board and investigated after written, informed consent was obtained. Separate consents were obtained for the skin biopsies, performed on patient 42 (19, 20) and patient 87. Patient numbers correspond to a master file of all NICHD patients with HPS. Control fibroblasts were purchased from the Coriell Human Genetic Mutant Cell Repository (Camden, NJ, U.S.A.) or were previously obtained from individuals not having HPS. Primary cultures of skin fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum containing 100 U/mL penicillin and 0.1 mg/mL streptomycin. For inhibition of nonsense-mediated mRNA decay, G-418 (Sigma Chemical-Aldrich, St. Louis, MO, U.S.A.), at a concentration of 200 μg/mL, was placed in the medium for 24 h before harvesting and RNA extraction.
Platelet whole mount electron microscopy.
Acid citrate dextran blood from the patient and an unaffected individual were centrifuged at temperatures between 24°C and 28°C at 5000 ×g for 20 min. The resulting platelet-rich plasma was deposited in 5-μL droplets onto carbon-coated Formvar grids, rinsed, and allowed to air dry as described (28). The grids were viewed and photographed with a Philips CM 10 (Philips, Eindhoven, The Netherlands) transmission electron microscope.
PCR amplification and sequencing.
Standard PCR procedures were used (29), using 58°C as the annealing temperature for all primers. Automated sequencing was performed on a Beckman CEQ 2000, using the CEQ Dye Terminator Cycle Sequencing kit according to the manufacturer's protocols (Beckman Coulter, Fullerton, CA, U.S.A.).
Northern blot analysis.
Total RNA from cultured fibroblasts was isolated by using the Trizol reagent (GIBCO BRL-Life Technologies, Gaithersburg, MD, U.S.A.), according to the manufacturer's guidelines. RNA (20 μg) was loaded in each lane of a 1.2% agarose/3% formaldehyde gel, which was electrophoresed and blotted onto a Nytran nylon membrane (Schleicher & Schuell, Keene, NH, U.S.A.) in the presence of 20 × SSC. The blot was prehybridized and then hybridized with ExpressHyb solution (Clontech, Palo Alto, CA, U.S.A.) at 68°C. The probe was human β3A cDNA, random-primer labeled with α[32P]dCTP (PerkinElmer Life Science, Boston, MA, U.S.A.) and prepared by PCR amplification of bp 1196–2424 of β3A cDNA (GenBank U91931); the β-actin probe was obtained from Clontech. After hybridization, the blot was washed three times with 2 × SSC/0.05% (wt/vol) SDS for 15 min at room temperature, and then twice with 0.1 × SSC/0.1% (wt/vol) SDS for 20 min at 50°C. The blot was then exposed to Kodak Biomax MR film, with an intensifying screen, for 24–72 h at −70°C.
Western blot analysis.
Protein extracts (20 μg) of cultured fibroblasts were electrophoresed on a 4–20% polyacrylamide gel (Bio-Rad, Hercules, CA, U.S.A.) and electroblotted onto 0.45-mm nitrocellulose membranes (Osmonics, Minnetonka, MN, U.S.A.). The blots were incubated with antibodies to the AP-3 subunits and to LAMP-1. Antigen-antibody complexes were detected using the enhanced chemiluminescence method (Amersham Pharmacia Biotech, Inc., Piscataway, NJ, U.S.A.) with horseradish peroxidase as detection ligand (Amersham Pharmacia Biotech, Inc.). Mouse monoclonal antiserum to LAMP-1 was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA, U.S.A.). Mouse monoclonal antiserum to μ3 (p47A) and ς3A were obtained from Transduction Laboratories (Lexington, KY, U.S.A.), and rabbit polyclonal antiserum to the β3A and δ3 subunits of AP-3 were kindly provided by A. Theos and M.S. Robinson (Cambridge Institute for Medical Research, Cambridge, U.K.).
Immunofluorescence and antibody internalization.
For steady state immunofluorescence (endogenous protein distribution), fibroblasts were grown on glass coverslips and fixed for 10 min in 2% formaldehyde in PBS. The cells were then incubated for 1 h with mouse MAb to LAMP-3 (Developmental Studies Hybridoma Bank) diluted in PBS, 0.2% saponin (wt/vol), and 0.1% BSA (wt/vol). After the incubation, unbound antibodies were removed by washing three times with PBS for 5 min. Cells were then incubated for 1 h with Cy-2 conjugated donkey anti-mouse secondary antibody (Jackson Immunoresearch, West Grove, PA, U.S.A.) also diluted in PBS/saponin/BSA. After washing the cells three times for 5 min in PBS, the cells were mounted onto glass slides with Fluoromont G (Southern Biotechnology Associates, Birmingham, AL, U.S.A.).
For antibody internalization studies, fibroblasts were grown on coverslips and incubated for 15 min at 37°C in the presence of LAMP-3 antibodies diluted in DMEM containing 0.1% (wt/vol) BSA, and 25 mM HEPES buffer, pH 7.4. Subsequently, cells were washed in ice-cold PBS, fixed in 2% formaldehyde for 10 min, and incubated for 1 h in the secondary antibody Cy-2 conjugated donkey anti-mouse, and diluted in PBS, 0.1% BSA, and 0.1% saponin. After washing the cells in PBS, the coverslips were mounted onto glass slides with Fluoromont G. Microscopy was performed on a Zeiss Axioscop-20 with a Cy-2 filter (Carl Zeiss, Inc., Welwyn Garden City, U.K.).
CASE REPORT
Patient 87 is a 5-y-old boy delivered to a 23-y-old woman who is three-fourths Cajun and one-fourth native American, i.e. Houma Indian. The father is believed to be half African American, one-fourth Chitimachi Indian, and one-fourth Houma Indian. The patient was born at full term via cesarean section due to footling breach presentation and a nuchal cord. Birth weight was 4.3 kg. Horizontal nystagmus and sparse white hair were present at birth. The hair darkened somewhat over time. He was circumcised without difficulty.
The first year of life was notable for several episodes of emesis, aspiration, and pneumonitis. Gastroesophageal reflux was demonstrated by pH probe. A severe respiratory syncytial virus infection required hospitalization for respiratory distress. There was also an episode of gross melena, associated with a rotavirus infection; packed red cells were administered. A brainstem auditory evoked response indicated a right-sided hearing loss. Complete blood cell counts were normal on several occasions except for a mild hypochromic, microcytic anemia. The white blood cell count at 7 mo of age was 10.9 × 103/μL with an absolute neutrophil count of 1635/μL. The bleeding time was 4 min. A sweat test and plasma amino acids were normal.
At 14 mo of age, the patient presented with severe respiratory distress, an influenza A virus infection, and hepatosplenomegaly. Admission weight was 7.4 kg, <3% for age. A complete blood cell count revealed a white blood cell count of 2.2 × 103/μL with an absolute neutrophil count of 1584/μL, Hb of 8 g/dL, and a platelet count of 288 × 103/μL. During a 3-mo hospitalization, a blood culture was positive for Enterococcus and a tracheal aspirate revealed Pseudomonas. Mechanical ventilation was required for more than 2 mo for acute respiratory distress syndrome, and there were four pneumothoraces. After a barium swallow revealed aspiration, a Nissen fundoplication procedure was performed, and a gastrostomy tube was placed for enteral feeding. The patient was discharged on oxygen by nasal cannula and has been using it ever since.
At age 18 mo, an immunologic investigation revealed that the IgA was 164 mg/dL (normal 16–95 mg/dL), the IgM 31 mg/dL (normal, 29–179 mg/dL), the IgG-1 646 mg/dL (normal, 170–950 mg/dL), the IgG-2 103 mg/dL (normal, 22–440 mg/dL), the IgG-3 78 mg/dL (normal, 4–69 mg/dL), and the IgG-4 18 mg/dL (normal, 0–20 mg/dL). The complement CH50 was 232 U/mL (normal, 100–250 U/mL). The diphtheria antibody titer was <1:40, but the tetanus antibody titer was 1:1280. The antinuclear antigen was negative.
At 20 mo of age, the patient had an additional episode of severe respiratory distress requiring mechanical ventilation and complicated by two pneumothoraces. Lung biopsy showed nonspecific interstitial pneumonitis. Long-term antibiotics and corticosteroids were administered.
At age 25 mo, the patient was again admitted for severe respiratory distress. The white blood cell count was 5.3 × 103/μL with 1% neutrophils, 92% lymphocytes, and 7% monocytes. The Hb was 10.1 g/dL, hematocrit 28.7%, and platelet count 628 × 103/μL. Daily G-CSF (Neupogen, Amgen, Inc., Thousand Oaks, CA, U.S.A.) increased the absolute neutrophil count to 12.6 × 103/μL within 4 d. Discontinuation of the G-CSF resulted in profound neutropenia with no cycling and repeated respiratory tract infections. Since that time, the patient has received 100 μg of G-CSF twice weekly. The nadir of the absolute neutrophil count has typically been 1100/μL, and the patient has not experienced any additional serious infections. His sole medications have been G-CSF, vitamins, and oxygen.
Lymphocyte types were enumerated while the patient was being treated with G-CSF. The white count was 24.3 × 103/μL with 7.29 × 103/μL lymphocytes, including 64% CD3, 16% CD4, and 48% CD8 cells. Thus, the percentage of CD4 cells was low, as was the CD4/CD8 ratio (0.33), but the absolute number of CD4 cells, 1166/μL, was normal. Since that time, the patient has had chicken pox, has received a live varicella virus immunization with a 1-mo postimmunization varicella IgG titer of 5.5 (indicative of past infection), and has tolerated immunization with live measles/mumps/rubella vaccine.
At 4 y of age, the patient had a platelet count of 378 × 103/μL and a bleeding time of >18 min. Mouth trauma caused bleeding for 24 h, stopped by platelet transfusions. Platelet aggregation studies showed a decreased response to epinephrine, collagen, and ADP. A liver biopsy for hepatomegaly revealed hepatocyte swelling, but electron microscopy did not demonstrate a storage disease or viral particles.
Magnetic resonance imaging of the head at 7 mo of age showed mild atrophy of the left temporal zone of the frontal lobe, consistent with severe chronic disease, and development progressed slowly. At 13 mo, the infant responded to voice but could not say words, sit, pull up, or walk. He rolled from back to front, but not from front to back. Over the next months, he began to sit alone and was scooting by age 18 mo. He walked and put words together at age 3 y and began scribbling at age 4. At age 5, he was able to speak full sentences, recite the alphabet, count to 20, run and jump, but was not toilet trained.
The patient had mild to moderate tricuspid valve regurgitation on echocardiogram. Pulmonary hypertension, with right ventricular and pulmonary artery pressures of 56.5 mm Hg, resolved by age 6.
On admission to the National Institutes of Health Clinical Center at 5 y of age, the patient was alert and active with a height of 88 cm (50% for a 2-y-old) and a weight of 13.1 kg (50% for a 2.3-y-old). Blood pressure was 110/68 mm Hg, and the heart rate was 109/min. The respiratory rate was 64/min and the patient was receiving 1–2 L/min of oxygen by nasal prongs. Other medications included cisapride 2.5 mg four times daily, folic acid 1 mg four times daily, sulfamethoxazole 200 mg twice daily, and trimethoprim 40 mg twice daily.
The cranium was normocephalic. The face was dysmorphic with epicanthal folds and slightly low-set and posteriorly rotated ears (Fig. 1A). The nasal root was broad, the nares prominent, the philtrum long, and the upper lip thin. Retrognathia was noticeable. The palate and uvula were normal. A single permanent upper central incisor was present, along with other deciduous teeth that were filled or crowned. Skin examination showed healed scars with no petechiae or ecchymoses. The hair was silvery blonde, and the eyes were green with a tan tint. Pendular horizontal nystagmus was noted. Cards were recognized at 3–4 feet. The fundus was blonde. A mild, 20–30 decibel, conductive hearing loss was ascertained bilaterally.

Clinical and laboratory findings in patient 87. (A) Lateral view of the face, showing long philtrum, micrognathia, slightly low-set and posteriorly rotated ears, light tan hair, and clubbing of the fingers. (B) Total body photograph showing short stature, genu valgus and tube placements. (C) Normal iris showing no transillumination. The normal contingent of pigment blocks light transmission. (D) Iris transillumination in the patient, indicating paucity of iris pigment. (Courtesy of Dr. M.I. Kaiser-Kupfer and E. Kuehl, National Eye Institute.) (E) Whole mount electron micrograph of dense bodies (black dots) in normal platelets (×1600). (F) Whole mount electron micrograph showing lack of dense bodies in patient's platelets (×1600).
Chest examination revealed a marked pectus deformity of the low anterior ribs bilaterally. Coarse breath sounds and scattered crackles were heard, and clubbing was present in the fingers (Fig. 1A). The heart examination was normal, and femoral pulses were strong. The abdomen was nontender but protuberant, with the liver 4 cm below the right costal margin and spleen 3 cm below the left costal margin. A gastrostomy tube was in place for delivery of nocturnal feedings (Fig. 1B). The musculoskeletal system was normal except for a valgus deformity of the knees.
Laboratory studies revealed a 46XY karyotype with normal fluorescent in situ hybridization (FISH) of a Williams syndrome probe to both #7 chromosomes. FISH studies for Prader-Willi and Angelman syndromes had previously been normal. Serum electrolytes, liver function tests, mineral studies, PTH, vitamin D, thyroid studies, immunoglobulins, sedimentation rate, and coagulation studies were normal. Total cholesterol was 170 mg/dL (normal, 100–200 mg/dL), with an HDL cholesterol of 78 mg/dL and LDL cholesterol of 55 mg/dL (normal, 65–129 mg/dL). The triglyceride concentration was 186 mg/dL. The Hb was 12.2 g/dL (normal, 11–15 g/dL) with a hematocrit of 37.0% (normal, 33–42%). Platelet counts were 428 and 426 × 103/μL (normal, 154–345 × 103/μL). The white blood cell count before a biweekly injection of G-CSF was 8.9 × 103/μL (43% polymorphonuclear leukocytes, 50% lymphocytes, 5% monocytes). The white blood cell count on the morning after a nighttime injection of G-CSF was 20.2 × 103/μL (71% polymorphonuclear leukocytes, 24% lymphocytes, and 4% monocytes). Lymphocyte phenotyping again revealed a low CD4/CD8 ratio of 0.50 (normal, 0.91–5.31). Oxygen saturation varied from 92 to 99% on room air.
Urinalysis was normal, with a specific gravity of 1.027. Daily urinary protein excretion was 23 mg; there was no glucosuria. Fractional excretion of phosphate was 2.2%. Creatinine clearance was 48 mL/min or 131 mL/min/1.73 m2. Radiographic results included a bone age of 3.5 y at a chronological age of 5.0 y; accessory epiphyses were noted in the heads of the second through fourth metacarpals. Spine radiographs were normal except for minimal scoliosis. Chest radiograph showed diffuse interstitial fibrosis, and chest computed tomography revealed bilateral upper lobe infiltrates and scattered scarring compatible with inflammatory disease. Recording of visual evoked potentials showed an asymmetric pattern typical of patients with albinism. A skin biopsy was performed and fibroblast cultures were established.
RESULTS
Laboratory diagnosis.
A bone marrow aspirate was performed while the patient was receiving G-CSF (Fig. 1C). Many mature neutrophils had Dohle bodies, a feature that was not noted before treatment with G-CSF. The mature neutrophils also lacked the toxic granulations characteristically seen after G-CSF administration. The giant intracellular inclusions characteristic of Chediak-Higashi syndrome (30) were absent from the myeloid series of cells on both Wright-Giemsa staining (Fig. 1C) and myeloperoxidase staining.
The diagnosis of HPS was supported by the finding of iris transillumination bilaterally (Fig. 1D), and was confirmed by wet mount electron microscopy of the patient's platelets showing absent dense bodies (28) (Fig. 1, E and F ).
Screening for mutations in HPS-causing genes.
As part of a routine protocol, the genomic DNA of patient 87 was sequenced exon by exon for mutations in the HPS1 gene, and no mutations were discovered. Next, the patient's RNA was examined for abnormalities in the size or amount of the ADTB3A transcript by Northern blot analysis. Patient RNA extracted from routinely cultured fibroblasts revealed complete absence of ADTB3A mRNA (Fig. 2). Verification of the lack of expression of the ADTB3A gene product was provided by Western blot analysis of protein extracted from the patient's fibroblasts. Patient 87 had no visible β3A protein and also completely lacked another AP-3 subunit, μ3 (Fig. 3). By comparison, fibroblasts from patient 42, one of the previously reported brothers with compound heterozygosity for in-frame mutations in ADTB3A(19, 20), had reduced but significant amounts of β3A and μ3 subunits, consistent with the normal contingent of β3A mRNA (Fig. 2). The reduction in the amount of ς3 and δ3 subunits was greater for patient 42 than for our current patient, 87 (Fig. 3).

Northern blot analysis of β3A expression. Total RNA extracts (20 μg) of three normal individuals (N-1, N-2, N-3) and of patient 42 and patient 87 were separated on a formaldehyde gel, blotted to a nylon membrane, and hybridized with α[32P]dCTP-labeled DNA probe to β3A (upper panel) and β-actin (lower panel), which served as a control.

Western blot analysis of AP-3 subunits. Protein extracts (25 μg) of cultured fibroblasts were electrophoresed on a 4–20% polyacrylamide gel, blotted onto nitrocellulose, and treated with antibodies to β3A, μ3, ς3A, and δ3, as well as LAMP-1, a control lysosomal membrane protein. Normal cell extracts from three different individuals are shown (N-1, N-2, N-3). Patient 42 is the previously described β3A-deficient individual (19, 20). Patient 87 is the child described in this article. Expression of β3A, μ3, ς3A, and μ3 were all decreased in patient 42, whereas β3A and μ3 were absent and ς3A was mildly deficient in patient 87. LAMP-1 protein levels were normal in each patient's fibroblasts, indicating that comparable amounts of protein were loaded in each lane.
Organization of the ADTB3A gene.
To perform mutation analysis on our patient's genomic DNA, we determined the organization of the ADTB3A gene. Three overlapping BACs (GI:12545290, 12583767, and 11863029) covered the 3′ portion of the gene, and the 14 exons and 13 introns in this region were identified by BLAST analysis (information available on the World Wide Web at http://www.ncbi.nlm.nih.gov/BLAST/). No genomic sequence was available for the 5′ end of the gene, so we treated the patient's fibroblasts with G-418, which can allow for correction of nonsense-mediated mRNA decay. Using this technique, we were able to prepare ADTB3A cDNA, including the first part of the gene. To determine the intron/exon boundaries of the first 13 exons of ADTB3A, we used the intron/exon boundaries in the homologous mouse gene, Ap3b1(31). The results indicate that human ADTB3A has 27 exons and 26 introns (Table 1).
Mutation analysis of genomic DNA.
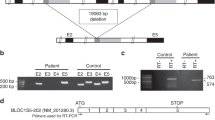
Using the newly determined exon/intron sequences of ADTB3A, exons 14–27 of the genomic DNA of patient 87 were amplified and sequenced. All had a normal sequence except exons 15 and 18. In exon 15 of patient 87 was found a heterozygous C1578T (R→X) mutation resulting in termination at codon 509 (Fig. 4). In exon 18 of patient 87 was present a heterozygous G2028T (E→X) mutation causing termination at codon 659 (Fig. 4). Genomic DNA of the patient's mother revealed that she was heterozygous for the C1578T mutation but not the G2028T mutation, indicating that the proband's mutations reside on separate alleles. Sequencing of cDNA obtained from G-418-treated fibroblasts verified the presence of the compound heterozygous mutations in patient 87. Paternal DNA was not available.

Mutation analysis of the ADTB3A gene in patient 87 and his mother. In exon 15, the patient and his mother each has a C1578T mutation at codon 509, changing a CGA (Arg) to a TGA (stop). In exon 18, patient 87 has a G2028T mutation at codon 659, changing a GAA (Glu) to a TAA (stop), whereas his mother has the normal sequence.
Cell biology.
Deficiency of the β3A subunit of AP-3 causes an increased trafficking of certain lysosomal membrane proteins through the plasma membrane of affected fibroblasts (19). One such lysosomal membrane protein is CD63 or LAMP-3. The fibroblasts of patient 87 have a normal contingent of intracellular and plasma membrane LAMP-3 when measured by direct antibody detection using permeabilized cells (Fig. 5A). This evaluates the steady-state level of LAMP-3 within the fibroblasts, because the LAMP-3 protein is freely accessible to the LAMP-3 antibody. In contrast, when nonpermeabilized cells are studied, the detection of LAMP-3 within cells requires antibody binding at the cell surface, followed by uptake into the cells. In these “internalization” experiments, nonpermeabilized cells are exposed to LAMP-3 antibody for 15 min. The total cell fluorescence provides a measure of how much LAMP-3 protein has circulated to the cell surface every 15 min, bound to LAMP-3 antibodies, and undergone internalization as an antigen-antibody complex subject to detection by indirect immunofluorescence. The increased accumulation of LAMP-3 antibody in the fibroblasts of patient 87 (Fig. 5B) indicates increased trafficking of LAMP-3 protein to the plasma membrane. Note the smaller increase in LAMP-3 trafficking to the plasma membrane in fibroblasts from patient 42, who produces residual amounts of β3A (19).

Internalization of LAMP-3 by normal and β3A-deficient fibroblasts. (Panel A) Endogenous LAMP-3 distribution, detected by indirect immunofluorescence using LAMP-3 antibody and permeabilized fibroblasts, in a normal individual (N), patient 42, and patient 87. (Panel B) LAMP-3 distribution in nonpermeabilized cells exposed to LAMP-3 antibodies for 15 min at 37°C. Indirect immunofluorescence indicates internalization of the LAMP-3 antigen-antibody complex, which in turn reflects trafficking of LAMP-3 protein to the cell surface. Endogenous LAMP-3 distribution, in a granular/lysosomal pattern throughout the whole cell, is similar in all three cell lines tested. The AP-3-deficient cell lines both show increased amounts of internalized LAMP-3 antibody when compared with the normal cell line, indicating increased trafficking of LAMP-3 protein to the cell surface. The effect is greater in cells from patient 87 than in cells of patient 42.
DISCUSSION
For decades, the enigmatic HPS was considered a disorder of membrane formation or breakdown affecting intracellular organelles such as the melanosome in melanocytes, the dense body of platelets, and the lysosome of various other cell types. In fact, these vesicles are related; they share at least one integral membrane protein. Specifically, ME491 in melanosomes, CD63 or granulophysin in platelets, and LAMP-3 in lysosomes are all the same protein (32–34). Moreover, melanosomes are considered to be of lysosomal lineage (35). Despite these considerations, the first direct evidence that HPS represents a membrane defect consisted of a report of the disorder in humans with a genetic deficiency of the β3A subunit of AP-3 (19).
At the time, AP-3 was already known to mediate the formation of intracellular vesicles from extant membranes, specifically, those of the trans-Golgi network. Clathrin binding by the heterotetrameric AP-3 complex has been demonstrated (23), and the preferences of its μ3 subunit for tyrosine and dileucine targeting signals has been defined (25, 26). In contrast, the particular type of vesicles that eventually form via the action of AP-3 was not determined. Nevertheless, the field of vesicular trafficking rapidly melded with that of human disease when the mouse pearl, a model of HPS, was found to have mutations in Ap3b1, the murine homolog of ADTB3A(27). The subsequent discovery of two human HPS patients with β3A deficiency (i.e. HPS-2) permanently linked the disease to defective vesicle formation/trafficking, and strongly suggested that other genetic causes of HPS, known and unknown, involve gene products responsible for membrane/vesicle interactions (4, 5).
Investigations of all three HPS-2 patients revealed important molecular, cellular, and clinical information about the disorder. The original two brothers with HPS-2 had compound heterozygous mutations in ADTB3A that allowed for some residual production of β3A. In contrast, patient 87 exhibited two nonsense mutations of ADTB3A, C1578T and G2028T, predicted to produce no residual β3A. The mutations were proven to be on separate alleles by investigation of the mother's genomic DNA; these studies included elucidation of the genomic organization of ADTB3A and its exon/intron boundaries (Table 1). Northern blots indicated a normal contingent of ADTB3A mRNA in patient 42 (Fig. 2), whereas patient 87 exhibited a complete absence of ADTB3A message, consistent with nonsense-mediated mRNA decay (36). Support for this mechanism was provided by the finding that treatment of the patient's fibroblasts with G-418 at least partially corrected the nonsense-mediated decay (37, 38), and allowed for enough mRNA production to permit reverse transcription of ADTB3A cDNA. Nonsense-mediated mRNA decay of two alleles of the same gene appears to be a rare occurrence.
The different levels of β3A expression in patients 42 and 87 had paradoxical effects on the endogenous levels of the other AP-3 subunits in cultured fibroblasts. Complete deficiency of β3A (in patient 87) resulted in complete lack of μ3, the AP-3 subunit to which β3A binds directly (24), and partial deficiency of β3A (in patient 42) significantly reduced μ3 levels. These findings are consistent with the hypothesis that β3A binding to μ3 stabilizes the complex against proteolytic degradation. The findings for δ3 and ς3A were unexpected, however. Fibroblasts with no β3A had nearly normal amounts of δ3 and ς3A, whereas cells from patient 42, with some residual β3A, displayed a marked reduction in δ3 and ς3A (Fig. 3). We speculate that, in the absence of β3A/μ3 complex formation, δ3 and ς3A cannot form a heterotetrameric complex and instead bind to each other to form a stable, protease-resistant complex. In contrast, when residual β3A/μ3 dyads containing mutated β3A subunits are available, the δ3 and ς3A subunits bind to them. However, the mutant β3A alters the conformation of the heterotetrameric complex sufficiently to render it susceptible to proteolytic degradation.
HPS-2 cells have also revealed information concerning the intracellular function of AP-3. AP-3-deficient melanocytes in culture have allowed determination of the role of AP-3 in targeting melanogenic proteins to the melanosome. Specifically, AP-3 was shown to mediate the trafficking of tyrosinase, but not TRP-1, to a melanosomal compartment (39). In fibroblasts, dysfunction of AP-3 results in increased plasma membrane concentrations of the lysosomal membrane proteins LAMP-1, LAMP-2, and CD63, or LAMP-3 (19). Apparently, disruption of the normal AP-3-mediated routing of these proteins allowed them to recycle through the plasma membrane as a default mechanism. The much more profound deficiency of AP-3 in patient 87 correlated with the pronounced accumulation of lysosomal membrane proteins on the surface of this patient's fibroblasts (Fig. 5).
Finally, the three HPS-2 patients provide the foundation upon which to craft a clinical description of this subtype of HPS. Children with ADTB3A mutations have oculocutaneous albinism, prolonged bleeding resulting from the absence of platelet dense bodies, a significant neutropenia responsive to G-CSF, a diathesis toward infections, and, perhaps, failure to thrive and dysmorphic features. Clearly, the most significant distinguishing feature of HPS-2 is neutropenia, and the relationship of this finding to AP-3 deficiency deserves further investigation.
The spectrum of HPS-2 disease, from mild in patients with residual β3A production to severe in a boy with no β3A, recapitulates the situation in the mouse. The pearl mouse, due to homozygous mutations allowing for residual β3A, displays a mild phenotype, with light pigment remaining throughout the hair shafts. In contrast, when the entire Ap3bI gene coding for β3A is knocked out by homologous recombination, the resulting mouse has more severe coat hypopigmentation and lacks pigment in its hair shafts (40). Variations in cellular manifestations of the ADTB3A defects can be studied using human cells cultured from patients with various degrees of residual β3A. In this fashion, the role of AP-3 in cargo recognition and vesicle formation can be further elucidated.
Patient 87 expands the phenotype of HPS-2 and also suggests that a broad spectrum of this disease may exist. The diagnosis should be considered in children with mild oculocutaneous albinism, a bleeding disorder, neutropenia, and an increased frequency of infections. In all three known HPS-2 patients, the diagnosis of Chediak-Higashi disease, characterized by oculocutaneous albinism, a platelet storage pool deficiency, giant intracellular inclusions, and a fatal infectious diathesis (30), was pursued. Clinicians might also suspect HPS-2 in children with the combination of oculocutaneous albinism, hematologic abnormalities, and minor dysmorphic features, as reported by Kotzot et al.(41). It will be of critical importance to describe in detail future HPS-2 patients, to accurately define the expression, course, and prognosis of this disease.
Abbreviations
- HPS:
-
Hermansky-Pudlak syndrome
- LAMP:
-
lysosome-associated membrane protein
- AP-3:
-
adaptor complex-3
- G-CSF:
-
granulocyte colony stimulating factor
References
Hermansky F, Pudlak P 1959 Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood 14: 162–169
Witkop CJ, Quevedo WC, Fitzpatrick TB, King RA 1989 Albinism. In: Scriver CR, Beaudet AL, Sly WS, Valle DL (eds) The Metabolic Basis of Inherited Disease, 6th Ed, Vol 2. McGraw-Hill, New York, pp 2905–2947
Gahl WA, Brantly M, Kaiser-Kupfer MI, Iwata F, Hazelwood S, Shotelersuk V, Duffy LF, Kuehl EM, Troendle J, Bernardini I 1998 Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky-Pudlak syndrome). N Engl J Med 338: 1258–1264
Shotelersuk V, Gahl WA 1998 Hermansky-Pudlak syndrome: models for intracellular vesicle formation. Mol Genet Metab 65: 85–96
Huizing M, Anikster Y, Gahl WA 2000 Hermansky-Pudlak syndrome and related disorders of organelle formation. Traffic 1: 823–835
McNicol A, Israels SJ 1999 Platelet dense granules: structure, function and implications for haemostasis. Thromb Res 95: 1–18
Hazelwood S, Shotelersuk V, Wildenberg SC, Chen D, Iwata F, Kaiser-Kupfer MI, White JG, King RA, Gahl WA 1997 Evidence for locus heterogeneity in Puerto Ricans with Hermansky-Pudlak syndrome. Am J Hum Genet 61: 1088–1094
Oh J, Ho L, Ala-Mello S, Amato D, Armstrong L, Bellucci S, Carakushansky G, Ellis JP, Fong C-T, Green JS, Heon E, Legius E, Levin AV, Nieuwenhuis HK, Pinckers A, Tamura N, Whiteford ML, Yamasaki H, Spritz RA 1998 Mutation analysis of patients with Hermansky-Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. Am J Hum Genet 62: 593–598
Oh J, Bailin T, Fukai K, Feng GH, Ho L, Mao J-I, Frenk E, Tamura N, Spritz RA 1996 Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet 14: 300–306
Bailin T, Oh J, Feng GH, Fukai K, Spritz RA 1997 Organization and nucleotide sequence of the human Hermansky-Pudlak syndrome (HPS) gene. J Invest Dermatol 108: 923–927
Dell'Angelica E, Aguilar R, Wolins N, Hazelwood S, Gahl WA, Bonifacino JS 2000 Molecular characterization of the protein encoded by the Hermansky-Pudlak Syndrome type 1 gene. J Biol Chem 275: 1300–1306
Oh J, Liu ZX, Feng GH, Raposo G, Spritz RA 2000 The Hermansky-Pudlak syndrome (HPS) protein is part of a high molecular weight complex involved in biogenesis of early melanosomes. Hum Mol Genet 9: 375–385
Witkop CJ, Babcock MN, Rao GHR, Gaudier F, Summers CG, Shanahan F, Harmon KR, Townsend DW, Sedano HO, King RA, Cal SX, White JG 1990 Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R 82: 333–339
Swank RT, Novak EK, McGarry MP, Rusiniak ME, Feng L 1998 Mouse models of Hermansky Pudlak syndrome: a review. Pigment Cell Res 11: 60–80
Wilson SM, Yip R, Swing DA, O'Sullivan TN, Zhang Y, Novak E, Swank RT, Russell LB, Copeland NG, Jenkins NA 2000 A mutation in Rab27a causes the vesicle transport defects observed in ashen mice. Proc Natl Acad Sci U S A 97: 7933–7938
Gardner JM, Wildenberg SC, Keiper NM, Novak EK, Rusiniak ME, Swank RT, Puri N, Finger JN, Hagiwara N, Lehman AL, Gales TL, Bayer ME, King RA, Brilliant MH 1997 The mouse pale ear (ep) mutation is the homologue of human Hermansky-Pudlak syndrome. Proc Natl Acad Sci U S A 94: 9238–9243
Feng GH, Bailin T, Oh J, Spritz RA 1997 Mouse pale ear (ep) is homologous to human Hermansky-Pudlak syndrome and contains a rare ‘AT-AC’ intron. Hum Mol Genet 6: 793–979
Spritz RA 2000 Hermansky-Pudlak syndrome and pale ear: melanosome-making for the millennium. Pigment Cell Res 13: 15–20
Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS 1999 Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol Cell 3: 11–21
Shotelersuk V, Dell'Angelica EC, Hartnell L, Bonifacino JS, Gahl WA 2000 A new variant of Hermansky-Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med 108: 423–427
Dell'Angelica EC, Ooi CE, Bonifacino JS 1997 β3A-adaptin, a subunit of the adaptor-like complex AP-3. J Biol Chem 272: 15078–15084
Schekman R, Orci L 1996 Coat proteins and vesicle budding. Science 271: 1526–1533
Dell'Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS 1998 Association of the AP-3 adaptor complex with clathrin. Science 280: 431–434
Simpson F, Peden AA, Christopoulou L, Robinson MS 1997 Characterization of the adaptor-related protein complex, AP-3. J Cell Biol 137: 835–845
Ohno H, Fournier MC, Poy G, Bonifacino JS 1996 Structural determinants of interaction of tyrosine-based sorting signals with the adaptor medium chains. J Biol Chem 271: 29009–29015
Hönig S, Sandoval IV, von Figura K 1998 A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosine mediates selective binding of AP-3. EMBO J 17: 1304–1314
Feng L, Seymour AB, Jiang S, To A, Peden AA, Novak EK, Zhen L, Rusiniak ME, Eicher EM, Robinson MS, Gorin MB, Swank RT 1999 The β3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky-Pudlak syndrome and night blindness. Hum Mol Genet 8: 323–330
Witkop CJ, Krumwiede M, Sedano H, White JG 1987 Reliability of absent platelet dense bodies as a diagnostic criterion for Hermansky-Pudlak syndrome. Am J Hematol 26: 305–311
Sambrook J, Fritsch EF, Maniatis T 1989 Molecular Cloning: A Laboratory Manual, 2nd Ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Introne W, Boissy RE, Gahl WA 1999 Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab 68: 283–303
Feng L, Rigatti BW, Novak EK, Gorin MB, Swank RT 2000 Genomic structure of the mouse Ap3b1 gene in normal and pearl mice. Genomics 69: 370–379
Hotta H, Ross AH, Huebner K, Isobe M, Wendeborn S, Chao MV, Ricciardi RP, Tsujimoto Y, Croce CM, Koprowski H 1988 Molecular cloning and characterization of an antigen associated with early stages of melanoma tumor progression. Cancer Res 48: 2955–2962
Nishibori M, Cham B, McNicol A, Shalev A, Jain N, Gerrard JM 1993 The protein CD63 is in platelet dense granules, is deficient in a patient with Hermansky-Pudlak Syndrome, and appears identical to granulophysin. J Clin Invest 91: 1775–1782
Hunziker W, Geuze HJ 1996 Intracellular trafficking of lysosomal membrane proteins. Bioessays 18: 379–389
Orlow SJ 1995 Melanosomes are specialized members of the lysosomal lineage of organelles. J Invest Dermatol 105: 3–7
Frischmeyer PA, Dietz HC 1999 Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet 8: 1893–1900
Burke JF, Mogg AE 1985 Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res 13: 6265–6572
Howard M, Frizzell RA, Bedwell DM 1996 Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med 2: 467–469
Huizing M, Saranjarajan R, Strovel E, Zhao Y, Gahl WA, Boissy RE 2001 AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol Biol Cell 12: 2075–2085
Yang W, Li C, Ward DM, Kaplan J, Mansour SL 2000 Defective organellar membrane protein trafficking in Ap3b1-deficient cells. J Cell Sci 113: 4077–4086
Kotzot D, Richter K, Gierth-Fiebig K 1994 Oculocutaneous albinism, immunodeficiency, hematological disorders, and minor anomalies: a new autosomal recessive syndrome?. Am J Med Genet 50: 224–227
Acknowledgements
The authors thank Drs. Hans Andersson and Jan Schmidt for their advice and assistance, and Drs. Alex Theos and Margaret S. Robinson for the use of their AP-3 subunit antibodies. We also thank Charles Galanis and Catherine Stimets Koss for excellent work in screening with Western blots.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Huizing, M., Scher, C., Strovel, E. et al. Nonsense Mutations in ADTB3A Cause Complete Deficiency of the β3A Subunit of Adaptor Complex-3 and Severe Hermansky-Pudlak Syndrome Type 2. Pediatr Res 51, 150–158 (2002). https://doi.org/10.1203/00006450-200202000-00006
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200202000-00006
This article is cited by
-
Mutational spectrum of the AP3B1 gene in an Iraqi family affected with Hermansky–Pudlak syndrome type 2
Egyptian Journal of Medical Human Genetics (2023)
-
Hermansky-Pudlak syndrome and oculocutaneous albinism in Chinese children with pigmentation defects and easy bruising
Orphanet Journal of Rare Diseases (2019)
-
Current Updates on Classification, Diagnosis and Treatment of Hemophagocytic Lymphohistiocytosis (HLH)
The Indian Journal of Pediatrics (2016)
-
Synergistic defects of UNC13D and AP3B1 leading to adult hemophagocytic lymphohistiocytosis
International Journal of Hematology (2015)
-
Cellular stress pathways in pediatric bone marrow failure syndromes: many roads lead to neutropenia
Pediatric Research (2014)
