
Abstract
The COL9A3 gene encodes one of the three alpha chains of Type IX collagen, with heterozygous variants reported to cause multiple epiphyseal dysplasia, and suggested as contributory in some cases of sensorineural hearing loss. Patients with homozygous variants have midface hypoplasia, myopia, sensorineural hearing loss, epiphyseal changes and carry a diagnosis of Stickler syndrome. Variants in COL9A3 have not previously been reported to cause vitreoretinal degeneration and/or retinal detachments. This report describes two families with autosomal dominant inheritance and predominant features of peripheral vitreoretinal lattice degeneration and retinal detachment. Genomic sequencing revealed a heterozygous splice variant in COL9A3 [NG_016353.1(NM_001853.4):c.1107 + 1G>C, NC_000020.10(NM_001853.4):c.1107 + 1G>C, LRG1253t1] in Family 1, and a heterozygous missense variant [NG_016353.1(NM_001853.4):c.388G>A p.(Gly130Ser)] in Family 2, each segregating with disease. cDNA studies of the splice variant demonstrated an in-frame deletion in the COL2 domain, and the missense variant occurred in the COL3 domain, both indicating the critical role of Type IX collagen in the vitreous base of the eye.
Similar content being viewed by others


Introduction
COL9A3 (OMIM#120270; NM_001853.4) encodes one of the alpha chains of the Type IX collagen heterotrimeric molecule which is expressed in the vitreous humour, cartilage of the cochlea, joints and spine [1]. Type IX collagen belongs to the fibril-associated collagen with interrupted triple helix family and provides support in connective tissues [2]. Autosomal dominant inherited variants in COL9A3 have been identified in individuals with multiple epiphyseal dysplasia (MED) (OMIM:600969), and may also be associated with a sensorineural hearing loss phenotype [3,4,5,6].
Stickler syndrome (STL) is characterised by ophthalmic, orofacial, articular and auditory features including myopia and vitreous abnormalities, cleft palate and midface hypoplasia, precocious arthritis, and conductive or sensorineural hearing loss. There is clinical variability within and among families and the lack of consensus regarding minimal clinical diagnostic criteria [7]. To date, autosomal dominant forms of STL are reported due to the heterozygous variants in COL2A1 (OMIM#108300), COL11A1 (OMIM#604841) and COL11A2 (OMIM#184840), while autosomal recessive forms are reported less frequently with biallelic variants in genes COL9A1 (OMIM#614134), COL9A2 (OMIM#614284) and recently COL9A3 [8,9,10].
Variants in COL9A3 have not been reported with autosomal dominant STL or Stickler-like phenotype. Autosomal dominant STL frequently has a notable vitreous phenotype, with COL2A1 variants associated with a membranous vitreous anomaly or an afibrillar appearance, and COL11A1 variants associated with a beaded vitreous appearance [11]. In autosomal recessive STL due to COL9A1 variants, the vitreous abnormality resembles an aged vitreous rather than a membranous, afibrillar/beaded appearance [12]. The predominant ocular phenotype in autosomal recessive STL due to COL9A2 and COL9A3 variants is high myopia, with no vitreoretinal abnormalities reported in those with COL9A3 variants [10]. Here, we report two families with heterozygous COL9A3 variants as the cause of severe peripheral lattice vitreoretinal abnormalities, with mild/moderate sensorineural hearing loss in some cases.
Materials and methods
Refer to Supplementary Materials for methods concerning subjects, ophthalmic examination, genomics, bioinformatics and sequencing, and RNA splicing assay.
Results
Clinical phenotype of vitreoretinal degeneration
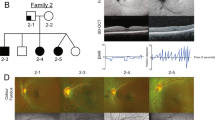
In Family 1, 14 affected individuals of Filipino/Australian ethnicity presented with vitreoretinal degeneration in a pattern suggestive of autosomal dominant inheritance (Fig. 1A). Affected individuals had extensive bilateral lattice vitreoretinal degeneration, with an abnormal vitreoretinal interface particularly at the vitreous base, where the retina was thinned and prone to tears. In Family 2 from New Zealand, three affected members of European background presented with vitreoretinal degeneration and retinal detachment, also in a pattern suggestive of autosomal dominant inheritance (Fig. 1B). In affected individuals in both families with extensive vitreoretinal degeneration, laser intervention or cryotherapy was recommended to prevent further vitreoretinal detachment or tearing (Fig. 1C–H). Further detailed clinical information is provided in Supplementary Materials: Clinical Information, and Table 1.

A, B Pedigrees showing affected individuals shaded in black. All affected individuals examined molecularly were heterozygous for the COL9A3:c.1107 + 1G>C variant in Family 1, or heterozygous for the COL9A3:c.388G>A in Family 2 (Detected by: *whole genome sequencing; +next-generation panel sequencing, #Sanger sequencing; ^wild-type alleles only). Grey shading indicates significant ocular phenotype, but individual not available for full ophthalmic analysis. C Family 1 III.15 highlighting inferior lattice retinal degeneration indicated by Δ, and atrophic retinal hole is indicated by short arrow. D Family 1 II.15, left eye, showing extensive treatment including cryotherapy shown in the region of Σ. Long arrows outline where laser retinal photocoagulation has been performed to areas of the retina that has lattice retinal degeneration. E, F Family 1 II.11, peripheral area of lattice retinal degeneration (area indicated by Δ) before and after retinal laser applied in barrier fashion. Short arrow indicates atrophic retinal hole within area of lattice retinal degeneration. Long arrows indicate areas where laser retinal photocoagulation has been performed. G Family 2 III.2, Optical coherence tomography (OCT) imaging of the right optic nerve and surrounding retina demonstrating significant traction at the vitreoretinal interface, with schitic and microcystic change in the ganglion cell layer, and outer retinal layers. H, I Retinal images of right and left eye of individual Family 2 III.2 showing thin myopic retina, and peripheral retinal barrier laser around retinal holes.
Genomics identifies COL9A3 variants in both families
In Family 1, whole genome sequencing identified no variants of significance in known autosomal dominant vitreoretinopathy disease genes. Detailed analysis looking for common alleles in six available affected family members, identified the heterozygous variant COL9A3:NC_000020.10(NM_001853.4):c.1107 + 1G>C, involving a canonical donor splice site within intron 21. This variant was absent from gnomAD and ClinVar databases. Predictive in silico modelling suggested the abolishment of the natural donor splice site and the skipping of exon 21 during RNA processing (Fig. 2A). Further investigations on whole blood RNA isolated from Patient II.13 showed that the essential donor splice site variant in COL9A3 induced skipping of exon 21 (Fig. 2A). This change resulted in the absence of 54 nucleotides from the COL9A3 cDNA (Fig. 2B), and the removal of 18 amino acids encoding 6 Glycine triplet repeats in the COL2 domain [COL9A3:NC_000020.10(NM_001853.4):c.1107 + 1G>C r.1054_1107del p.(Gly352_Pro369del)] (Fig. 2C).

A cDNA studies of the COL9A3 splice variant, showing neighbouring COL9A3 exon structure (exons are numbered as per NCBI RefSeq NG_016353.1), and the location of the c.1107 + 1G>C variant with an asterisk, and the predicted aberrant splicing event. Agarose gel image of RT-PCR products amplified from whole blood RNA depicting exon 21 skipping with two sized bands, the 290bp band from the normal allele and the 236bp band from the allele with the c.1107 + 1G>C variant present in the patient, and only the band from the normal allele present in the control. Pt1 patient 1 (Patient II.13, Family 1), Ctrl unaffected control. B Sanger sequencing confirmation of the skipping of exon 21 and subsequent deletion of 54bp from the mutant RNA sequence in Pt1, compared with wildtype RNA sequence in the control. C Protein structure diagram of COL9A3, labelled with amino acid numbering on functional domain boundaries. Non-collagen domains [NC1–4] and Collagen domains [COL1–3], respectively. Previously reported variants were sourced from the HGMD public database (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=COL9A3) [Accessed: April 2020]. Heterozygous variants associated with multiple epiphyseal dysplasia (MED) in purple; heterozygous variants associated with isolated hearing loss in green; heterozygous variants identified in this study are in red italics. An asterisk indicates change due to splicing variants.
In Family 2, connective tissue panel sequencing in Patient III.2 (CTGT, Pennsylvania, USA) identified the heterozygous missense variant in COL9A3:NC_000020.10(NM_001853.4):c.388G>A p.(Gly130Ser). This change predicts the substitution of a highly conserved Glycine to a Serine residue at amino acid position 130, located in the COL3 domain. This substitution is ascribed as probably damaging by PolyPhen2, and is present in gnomAD at an allele frequency of 0.0008, with multiple submissions as a variant of uncertain significance in ClinVar.
Segregation studies confirmed autosomal dominant inheritance in both families of the COL9A3 variants identified (Fig. 1A, B). Variants were submitted to the ClinVar database with accession numbers: SCV001424845 and SCV001424846.
Discussion
We have identified heterozygous COL9A3 variants in two families as the likely cause of a severe peripheral lattice vitreoretinal degeneration, with a strong propensity for subsequent retinal detachment. Importantly, the affected members of both families primarily presented with predominant ocular features of vitreoretinal degeneration and retinal detachment. Interestingly, most who had audiological evaluation were found to have mild/moderate sensorineural hearing loss suggesting a Stickler-like phenotype. This is the first report of cases with an autosomal dominant variant in a Type IX collagen gene leading to a severe predominantly vitreoretinal degenerative phenotype. To date, myopia has been the main ocular feature in cases with homozygous frameshift variants and extraocular features indicating a diagnosis of STL [8, 9], with a hypoplastic vitreous reported in one family [10] (Fig. 2C).
The Type IX collagen heterotrimer is comprised of collagenous domains (COL1–COL3) and non-collagenous domains (NC1–NC4) (Fig. 2C) [2]. Variant type and position involving collagen genes may have an association with disease type or severity, as noted in the Type II collagenopathies [13]. Variants affecting collagenous domains due to splice variants with in-frame deletions, or missense variants affecting glycine residues, as identified in the two families in this study, may have dominant-negative effects interfering with protein function [13, 14]. In Type II collagenopathies, these variant types may result in a more severe phenotype, compared with nonsense or frameshift variants which may lead to reduced synthesis of normal collagen, and milder phenotypes observed [13]. It is likely that in the Type IX collagenopathies, disease type or severity may also be influenced by the protein domain affected and the variant type.
In Family 1, we describe the first heterozygous in-frame deletion due to a splice site variant within the COL2 domain of the Type IX collagen alpha-3 chain (Fig. 2C). Other heterozygous in-frame deletions due to splice site variants in Type IX collagen alpha chains occur in the COL3 domain and lead to MED [3, 15, 16] (Fig. 2C), likely due to the importance of the COL3 domain in the cartilage growth plate [17]. Given the predominant ocular phenotype observed in Family 1 patients with a heterozygous COL2 domain in-frame deletion, this suggests a critical role for the COL2 domain of Type IX collagen in the vitreous. Type II and XI collagens form fibrils in the vitreous, and Type IX collagen is present in the outer sheaf-like conduit maintaining appropriate inter-fibril distance with chondroitin sulfate interactions [18]. Type II collagen is the predominant collagen in the vitreous and the collagen fibrils are at their highest concentration at the vitreous base, where they prevent the vitreous from separating from the retina. The integrity of the fibrils in this region is also maintained by interaction with Type IX collagen [18]. Type II collagen has been shown to bind to a region in the COL2 domain of COL9A3, which interestingly lies in close proximity to the in-frame deletion identified in our family [19]. The COL9A3 in-frame deletion in our family may disrupt the required Type II collagen and COL9A3 interaction at the vitreous base, through misalignment of the Type IX collagen chains. Misalignment due to heterozygous in-frame deletions with a dominant-negative effect on collagen function has been postulated as disease-causing in other collagenopathies [14].
The missense variant identified in Family 2 lies within the COL3 domain of Type IX collagen. The COL3 domain has been found to bind to the extracellular matrix protein Matrilin-3, important for collagenous tissue integrity, and therefore also of likely importance to vitreous integrity [17, 20].
The COL9A3:c.388G>A variant has a population allele frequency of 0.0008 in gnomAD, however it is unclear if vitreoretinal degeneration or retinal detachment cases are excluded from this database. Variable phenotypic expression has been observed in patients with MED due to the variation in COL9A3 [4]. While it is interesting to consider that variable expression of an ocular phenotype could also occur, it cannot be assumed that phenotypic variability would also apply for other conditions resulting from variants in the same gene. It is noteworthy however that the cumulative lifetime risk of retinal detachment may be in the vicinity of 0.03 even without a known family history, and there are likely other genetic and non-genetic factors that contribute to this risk [21]. Our study highlights that the missense variant identified in COL9A3 in Family 2 may lead to a vitreoretinal degeneration and detachment phenotype, and suggests that others with this variant in population databases may also be at increased risk of this phenotype.
This study is the first to describe autosomal dominant, heterozygous disease-causing variants in COL9A3 segregating with a striking peripheral vitreoretinopathy with resultant retinal detachment in almost all cases, and sensorineural hearing loss in some cases. This study highlights a likely critical role for COL9A3 in the function of Type IX collagen in its interaction with Type II collagen at the vitreous base. We propose COL9A3 should be examined in all pedigrees where bilateral vitreoretinal lattice degeneration and detachment are present, even in the absence of additional Stickler-like features.
References
Byron A, Humphries JD, Humphries MJ. Defining the extracellular matrix using proteomics. Int J Exp Path. 2013;94:75–92.
Kapyla J, Jaalinoja J, Tulla M, Ylostalo J, Nissinen L, Viitasalo T, et al. The fibril-associated collagen IX provides a novel mechanism for cell adhesion to cartilaginous matrix. J Biol Chem. 2004;279:51677–87.
Paassilta P, Lohiniva J, Annunen S, Bonaventure J, Le Merrer M, Pai L, et al. COL9A3: A third locus for multiple epiphyseal dysplasia. Am J Hum Genet. 1999;64:1036–44.
Nakashima E, Kitoh H, Maeda K, Haga N, Kosaki R, Mabuchi A, et al. Novel COL9A3 mutation in a family with multiple epiphyseal dysplasia. Am J Med Genet A. 2005;132A:181–4.
Asamura K, Abe S, Fukuoka H, Nakamura Y, Usami S. Mutation analysis of COL9A3, a gene highly expressed in the cochlea, in hearing loss patients. Auris Nasus Larynx. 2005;32:113–7.
Miyagawa M, Naito T, Nishio SY, Kamatani N, Usami S. Targeted exon sequencing successfully discovers rare causative genes and clarifies the molecular epidemiology of Japanese deafness patients. PLoS ONE. 2013;8:e71381.
Robin NH, Moran RT, Ala-Kokko L. Stickler syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington; 2017.
Faletra F, D’Adamo AP, Bruno I, Athanasakis E, Biskup S, Esposito L, et al. Autosomal recessive Stickler syndrome due to a loss of function mutation in the COL9A3 gene. Am J Med Genet A. 2014;164A:42–47.
Hanson-Kahn A, Li B, Cohn DH, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian G, et al. Autosomal recessive Stickler syndrome resulting from a COL9A3 mutation. Am J Med Genet A. 2018;176:2887–91.
Nixon TRW, Alexander P, Richards A, McNinch A, Bearcroft PWP, Cobben J, et al. Homozygous type IX collagen variants (COL9A1, COL9A2, and COL9A3) causing recessive Stickler syndrome—expanding the phenotype. Am J Med Gen A. 2019;179:1498–506.
Richards AJ, Baguley DM, Yates JR, Lane C, Nicol M, Harper PS, et al. Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the Type II collagen Gly-X-Y triple helix. Am J Hum Genet. 2000;67:1083–94.
Nikopoulos K, Schrauwen I, Simon M, Collin RW, Veckeneer M, Keymolen K, et al. Autosomal recessive Stickler syndrome in two families is caused by mutations in the COL9A1 gene. Invest Ophthalmol Vis Sci. 2011;52:4774–9.
Barat-Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D, et al. Mutation update for COL2A1 gene variants associated with type II Collagenopathies. Hum Mutat. 2016;37:7–15.
Griffith AJ, Sprunger LK, Sirko-Osada DA, Tiller GE, Meisler MH, Warman ML. Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am J Hum Genet. 1998;62:816–23.
Czarny-Ratajczak M, Lohiniva J, Rogala P, Kozlowski K, Perala M, Carter L, et al. A mutation in COL9A1 causes multiple epiphyseal dysplasia: Further evidence for locus heterogeneity. Am J Hum Genet. 2001;69:969–80.
Muragaki Y, Mariman EC, van Beersum SE, Perala M, van Mourik JBA, Warman ML, et al. A mutation in the gene encoding the alpha 2 chain of the fibril-associated collagen IX, COL9A2, causes multiple epiphyseal dysplasia (EDM2). Nat Genet. 1996;12:103–5.
Fresquet M, Jowitt TA, Ylostalo J, Coffey P, Meadows RS, Ala-Kokko L, et al. Structural and functional characterization of recombinant matrilin-3 A-domain and implications for human genetic bone diseases. J Biol Chem. 2007;282:34634–43.
Le Goff MM, Bishop PN. Adult vitreous structure and postnatal changes. Eye. 2008;22:1214–22.
Wu J, Woods PE, Eyre DR. Identification of cross-linking sites in bovine cartilage type IX collagen reveals an antiparallel typeii-type IX molecular relationship and type IX to type IX bonding. J Biol Chem. 1992;267:23007–14.
Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, et al. A subcellular map of the human proteome. Science. 2017;356:eaal3321.
Go SL, Hoyng CB, Klaver CCW. Genetic risk of rhegmatogenous retinal detachment: a familial aggregation study. Arch Ophthalmol. 2005;123:1237–41.
Acknowledgements
We would like to thank the families for their willingness to participate in this study. Support is acknowledged from the Ophthalmic Research Institute of Australia, the NSW Office of Health and Medical Research, Retina New Zealand, the Save Sight Society New Zealand, NHMRC Application 1116360 and the Sydney Eye Hospital Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nash, B.M., Watson, C.J.G., Hughes, E. et al. Heterozygous COL9A3 variants cause severe peripheral vitreoretinal degeneration and retinal detachment. Eur J Hum Genet 29, 881–886 (2021). https://doi.org/10.1038/s41431-021-00820-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00820-1
This article is cited by
-
Identification of three novel homozygous variants in COL9A3 causing autosomal recessive Stickler syndrome
Orphanet Journal of Rare Diseases (2022)
