
Abstract
Initial recognition of microbes, as they enter the body, is based on germ line–encoded pattern recognition receptors that selectively bind to essential components of pathogens. This allows the body to respond immediately to the microbial invasion before the development of active immunity. The signal-transducing receptors that trigger the acute inflammatory cascade have been elusive until very recently. On the basis of their genetic similarity to the Toll signaling pathway in Drosophila, mammalian Toll-like receptors (TLRs) have been identified. By now, nine transmembrane proteins in the TLR family have been described. Mammalian TLR4 is the signal-transducing receptor activated by the bacterial lipopolysaccharide. The activation of TLR4 leads to DNA binding of the transcription factor NF-κB, resulting in activation of the inflammatory cascade. Activation of other TLRs is likely to have similar consequences. TLR2 mediates the host response to Gram-positive bacteria and yeast. TLR1 and TLR6 may participate in the activation of macrophages by Gram-positive bacteria, whereas TLR9 appears to respond to a specific sequence of bacterial DNA. The TLRs that control the onset of an acute inflammatory response are critical antecedents for the development of adaptive acquired immunity. Genetic and developmental variation in the expression of microbial pattern recognition receptors may affect the individual's predisposition to infections in childhood and may contribute to susceptibility to severe neonatal inflammatory diseases, allergies, and autoimmune diseases.
Similar content being viewed by others

Main
Infections account for a significant proportion of morbidity in children, despite active immunization programs and the use of antibiotics. An acute inflammatory response in the absence of adaptive immune defense is essential for survival. An inappropriate innate immune response may lead to serious organ damage, allergies, or autoimmune diseases. A central question relevant to all mammals, and particularly germane to children, is the dynamic interplay between the host and potential pathogens.
Recent investigations led to the discovery of a class of cell surface–signaling molecules termed TLRs, which provide the missing link in the immune response. Several of them control the proinflammatory activation of macrophages and the onset of acute inflammatory cascades. It is becoming increasingly clear that innate immunity is more than the first line of host defense. It is actually a necessary antecedent for the development of an adaptive immune response. In this review, we will highlight the discovery of mammalian Toll receptors and speculate as to the potential implications of these findings for pediatric practices.
ACTIVATION OF ADAPTIVE IMMUNITY
The adaptive immune response is a critical component of the host's response to pathogens. The key features of adaptive immunity are the clonal expansion of lymphocytes in response to a particular antigen and the ability to evoke an immunologic memory. In the adaptive immune system, specific T- and B-cell receptors for each clone of cells respond to a specific antigen. Each clonal receptor in both B and T cells is structurally unique and not encoded in the germ line. A clonal receptor is not predestined to recognize any particular antigen, either. These antigen-specific receptors are not passed to the next generation and therefore have to be established by every generation, mostly during early childhood. This process is inadequate for protection of the young host. First, there is a lag period of more than a week for a primary response, or several days in a primed host, before a full adaptive immune response takes effect. The host's capability to limit the infection before adaptive immunity develops used to be a matter of life and death before the introduction of immunizations and antibiotics. Another critical issue is that the virtually limitless number of antigen receptors emerging as a result of random genetic mechanisms may potentially be capable of reacting not only with infectious microorganisms but also with various environmental antigens or even self-antigens. This may lead to allergies or autoimmune diseases.
INNATE IMMUNITY AND PATTERN RECOGNITION RECEPTORS AND MOLECULES
Innate immunity plays a primary role in acute host defense. It critically contributes to destroying the pathogen, determining the localization and extent of the challenge, and facilitating the adaptive immune response. Innate immunity, or first-line host defense, primarily serves to restrict the infectious challenge during the lag period required for adaptive immunity to develop. The innate component of immune recognition is based on a germ line–encoded recognition system. Pattern recognition receptors and molecules selectively recognize membrane-derived structures in pathogenic microorganisms. The latter include, among others, LPS (also called endotoxin) from the cell wall of Gram-negative bacteria, peptidoglycans, teichoic acids and lipoteichoic acid from Gram-positive bacteria, lipoglycan from Mycobacteria tuberculosis, and mannans from yeast and the outer surface proteins that adorn viruses.
The total number of receptors involved in innate recognition of antigens is limited to approximately 100. However, microbes are extremely heterogeneous and have very high mutation rates. The response of the innate immune system has been focused on the highly conserved and essential structures collectively present in many types of microorganisms. Many of the mammalian pattern recognition receptors date back hundreds of millions of years.
The absence of microbial markers on the surface of the host cells constitutes the basis of self versus nonself discrimination (1, 2). This requires receptor-ligand interaction between the host's pattern recognition molecules and specific molecular patterns in the microorganism. Although certain postreceptor events signal proliferation and differentiation of inflammatory cells, many host defense functions are activated instantaneously or at least within less than a day (1–3). The expression of the pattern recognition receptors is not clonal, and a given receptor expressed by a given cell type has a defined specificity to a certain group of microbes. The receptors of the innate immune system are preferentially expressed on monocytes and macrophages, but also on dendritic cells, B cells, and even in other cell types not traditionally considered in host defense.
Structurally, pattern recognition receptors have LRR domains, calcium-dependent lectin domains, or scavenger-receptor protein domains (1). Functionally, pattern recognition receptors can be divided into three classes labeled as secreted, endocytotic, or signaling molecules. The best-characterized secreted receptor is MBL, a member of the collagen-containing lectin or collectin family that includes SP-A, SP-D, and others. MBL is synthesized in liver and secreted into serum as a component of the acute-phase response. MBL binds to carbohydrates on Gram-positive and Gram-negative bacteria, yeast, and some other microorganisms. This leads to activation of MBL-associated proteases and consequent activation of the complement cascade by a mechanism not requiring antigen-antibody complexes (3).
Endocytic pattern recognition receptors occur on the surface of phagocytes. These receptors recognize the pathogen-associated molecular patterns and thereby move the pathogen into intracellular lysosomes of macrophages, where it is destroyed. Components of the pathogen may then be processed to be presented by major histocompatibility complex molecules on the surface of the macrophage. Macrophage mannose receptor and macrophage scavenger receptor are among the endocytic pattern recognition receptors identified thus far.
Signaling receptors recognize the pathogens and induce the expression of a variety of acute-phase reaction products, including the inflammatory cytokines. Receptors for these molecules are present in a variety of inflammatory and somatic cells and are capable of inducing a number of the mediators involved in acute host defense (the cytokine cascade). Some of the inducible molecules influence the presentation of the antigen to immunocompetent clonal cells. Epithelial cells, macrophages, natural killer cells, and antigen-presenting cells prominently line the mucous surfaces and express specific pattern recognition receptors. A long-term elusive problem has been the lack of knowledge of the signaling receptors that mediate the onset of the inflammatory cascade.
FAMILY OF TLR
The Toll gene was first described in the context of a pathway that prescribes the dorsal–ventral development in the Drosophila embryo (4). The detection of the link between this embryonic pathway and mammalian innate immunity is largely because of the recognition of the conserved pathways and motifs between insects and mammals. The similarity between the dif/dorsal pathway in Drosophila and the NF-κB pathway in mammals led to the realization that the Toll pathway also specifies antifungal responses at the later developmental stages of this insect (5). This study revealed remarkable homology in the cytoplasmic domains of two transmembrane proteins, namely Drosophila Toll and the mammalian IL-1RI, as shown in Figure 1 (6–9). These observations, in turn, linked the signal transduction pathways in Drosophila and mammals and led to the discovery of a human TLR (now termed TLR4). Ligation of TLR4 induced NF-κB activation, cytokine release, and induction of costimulatory molecules on macrophages that, in turn, assisted the activation of naïve T cells (6).

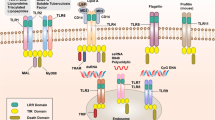
Schematic representation of the protein structure of human TLRs (6–9), Drosophila melanogaster Toll, and vertebrate IL-1RI. Individual 20- to 29-amino acid LRR are indicated as black circles. The ectodomain consisting of 18–24 LRR shows variation among different human TLRs and among different species. A sequence of approximately 60 amino acids, including two to four conserved cysteines, is shown as a white box. This domain is conserved in several other human proteins, such as platelet glycoprotein IX and oncofetal antigen 5T4 (Genebank accession numbers X52997 and N86494, respectively). A cysteine-rich domain of approximately 90 amino acids present in Drosophila Toll, but missing from human TLRs, is depicted as two white semicircles opposite to each other. The ectodomain of IL-1RI consists of three immunoglobulin-like domains. An intracellular signaling domain of approximately 200 amino acids common to all TLRs and vertebrate IL-1RI is indicated as a gray ellipse. This TLR/IL-1R or TIR domain is highly conserved among different TLRs and IL-1RI and among different animals.
To date, at least nine mammalian genes encoding mammalian TLR molecules have been identified (6–10). As depicted in Figure 1, TLRs are type 1 membrane proteins. The extracellular domain (ectodomain) of TLR contains LRR. All TLRs have a single transmembrane domain and a cytoplasmic domain involved in signal transduction. This cytoplasmic domain is referred to as the TIR domain on the basis of its homology with IL-1RI.
From DNA sequence comparisons of TLR genes among Drosophila, reptiles, birds, and mammals, the genes are exceptionally well conserved. A common ancestor gene may have been present >350 million years ago. Compared with the cytoplasmic domain, the extracellular LRR is less well conserved between, for instance, man and mouse.
HOW THE HOST USES TLRS
The host response to pathogenic microorganisms depends on recognition of the antigenic determinants of the pathogen and activation of specific effector mechanisms. These include the appropriate postreceptor events for the elimination of the pathogen, signaling as to the route of entry, and localization of the pathogen in the host. Adaptive immunity in vertebrates uses the innate system in the selection and presentation of antigens to clonotypic recognition systems.
In mammals, TLR transmits the signal from the ectodomain to the cytoplasm, leading ultimately to the activation of NF-κB [Fig. 2;(11–14)]. This process has been best described between LPS and TLR4. The LPS-induced activation of TLR4 is mediated via the TIR domain that uses a conserved signal transduction pathway. This pathway requires MyD88, IRAKs, and other downstream intermediate proteins, not shown in Figure 2 (15, 16). MyD88 is a protein that interacts with the TLRs through its own carboxy-terminal TIR domain. Through its amino-terminal death domain, MyD88 recruits IRAK to propagate the proinflammatory signal that ultimately phosphorylates the IκB kinase complex consisting of IκB kinase α and β and a scaffold protein NEMO/IKKγ(17–19). This phosphorylation event liberates bound NF-κB from the cytoplasm into the nucleus. Both NF-kB and the activator protein-1 family (c-Jun, fos, ATF2, ELK1, and others) serve as nuclear transcription factors (some factors affect the stability of mRNA) in this chain of events. By binding cognate cis elements in many genes that control inflammatory events, NF-κB induces IL-1, IL-6, TNF-α, inducible NO synthase, and others. Secondary activation of cytokine receptors, which are widely distributed among immune and nonimmune cells, induces further cascades of cytokines, cytokine antagonists, chemokines, adhesion molecules, vasoactive agents, oxidants, antioxidants, proteases, antiproteases, and stress hormones. They are involved in various facets of the host defense or up-regulate the stress resistance of the host.

The pathway for the innate immune response in insects and in mammals. The homology between insects and mammals is illustrated by the similar shapes of parts of the molecules. Otherwise, the shapes are arbitrary. The following homologies between insects and mammals are evident: Tube and MyD88, Pelle and IRAKs, Cactus and IκB, Dorsal and NF-κB, respectively. The Toll ligand Spätzle is likely to mediate both host defense and embryogenesis (11), whereas LPS activates TLR4 in mammals. LPS also interacts with extracellular CD14 and LPS-binding protein (LBP), and MD-2 is additionally required for activation of TLR4 (12–14). In mammals, TLR-4, other TLRs (not shown), IL-1RI, and IL-18 receptor (not shown) share a similar intracellular domain, called Toll/IL-1R domain or TIR domain (Fig.1). The TNF receptor (TNFR) is not a member of the TLR/IL-1R superfamily, and TNFR additionally signals apoptosis. Some of the downstream components in the inflammatory cascade are shown. Intracellular MyD88 interacts with TIR domains of TLR and IL-1R via its own TIR domain. MyD88 recruits IRAKs via their death domains (gray areas). Activation of TLR, IL-1, and TNFRs results in release into the nucleus of NF-κB and the NFs of the activator protein-1 (AP-1) family. They are the immediate activators of the inflammatory cascade that includes inflammatory cytokines, inducible NO synthase (NOS-2), and other mediators.
The Drosophila Toll pathway is critical for the regulation of drosomycin, an antifungal peptide that is synthesized in the fat body of the fly in response to a fungal challenge (11). Activation of Toll results in nuclear translocation of Dorsal, which is homologous to NF-κB in mammals. This then permits transcriptional up-regulation of the Drosophila drosomycin gene. Toll in Drosophila also serves as a dorsoventral embryonic regulatory signal, which is triggered by the Spätzle protein that is cleaved by a protease from its precursors (Fig. 2).
TOLL AGONISTS
The initial description of human TLR4 did not specify a physiologic ligand, although it was postulated that this would be a microbial cell wall product (6). The clue to this puzzle came from the long-standing observation that two mouse strains, C3H/HeJ and C57BL/10ScCr, were resistant to LPS (20, 21). Using a positional cloning approach, the locus was mapped to murine chromosome 4, the locus of the TLR4 gene that proved to be what was later called the LPS gene. A histidine-to-proline mutation was found in C3H/HeJ mice, and the entire TLR4 gene was deleted in C57BL/10ScCr mice (22). Animals with deleted TLR4 gene expression fail to respond to LPS. These observations and those on Chinese hamster cells support the assumption that TLR4 is an essential element of the LPS receptor complex that directs subcellular responses to LPS in mice and, presumably, in humans (12, 22, 23).
Whether microbial products bind directly to the macrophage-associated TLR remains uncertain. LPS first interacts with the serum LPS-binding protein, and activation of monocyte-macrophages takes place after LPS (or peptidoglycan) binds to macrophage CD14. The latter is a glycosylphosphatidylinositol-linked cell surface glycoprotein and a TLR homologue with a leucine-rich extracellular domain. However, CD14 lacks the cytoplasmic domain and thus does not transduce a signal into the cell. The absence of CD14 reduces rather than abolishes the LPS or peptidoglycan response (12, 13). It may be that CD14-microbe complexes bind to or interact with TLRs, which are present in low numbers [1000 or fewer molecules per macrophage;(24)]. Another novel molecule, MD-2, binds to LPS and confers the response of TLR4 to LPS. MD-2 may be the link between LPS and the signaling of TLR4 (14).
The density of TLR influences the degree of inflammatory response to a microbial toxin (12). Considering the similarities in pathogens among vertebrates, the large interspecies difference in the TLR4 extracellular domain is remarkable. This is consistent with the possibility that the variable LPS structures from different Gram-negative bacteria exert a selection pressure on the genetic structure of the molecule. In addition, differences among human individuals in the LPS responsiveness of macrophages have also been observed, and a patient refractory to LPS effects was reportedly predisposed to recurrent infections (25). According to preliminary evidence, a common human genetic variation affecting a single amino acid in the extracellular domain of TLR4 altered the responsiveness of the airways to LPS challenge (26).
On the basis of expression in heterologous cells in vitro, LPS was deemed to also interact with TLR2(27, 28). However, mice lacking TLR2 expression normally respond to LPS (29), but had a very low response to toxins from Gram-positive bacteria (30). Furthermore, TLR2 was recruited to macrophage phagosomes containing specifically Gram-positive bacteria or yeast, whereas TLR4 was recruited to phagosomes containing Gram-negative bacteria (12, 30, 31). TLR2 seems to be a receptor for components of Gram-positive bacteria, mycobacteria, and yeast. According to Ozinsky et al. (32), the cytoplasmic TIR domains of individual TLRs are not functionally equivalent. TLR2 and TLR6 were both recruited to the macrophage phagosome, where they recognized peptidoglycan from Gram-positive bacteria, whereas only TLR2 and another unidentified TLR were required to identify Gram-positive bacterial lipopeptide. When constitutionally activated, the TLR4 homodimer became functionally active. However, TLR2 required TLR6 or TLR1 pairs in order to be functionally equivalent with TLR4, as judged on the basis of TNF induction.
Nucleotide sequences containing unmethylated CpG are found in prokaryotic DNA at much higher frequencies than in vertebrate DNA. CpG induced production of immune-stimulating cytokines in macrophages, dendritic cells and natural killer cells (33). Hemmi et al. (34) recently demonstrated that TLR9-deficient mice showed no immune response to CpG DNA. This led the investigators to postulate that TLR9 is involved in immunogenic identification of bacterial DNA.
It has been suggested that the expressions of TLR2 and TLR4 are restricted to hematopoietic cells, although studies show wide tissue distribution [Table 1;(7–10, 35, 36)]. TLR4 was expressed in cardiac myocytes, in which ischemia induced its expression (35). The roles of the different members in the TLR family, identification of their respective ligands, and detailed characterization of the proteins recruited to their respective signalsomes remain to be determined.
Does the human TLR serve functions other than signaling a response to microbes? The first reports on possible endogenous ligands for TLRs are being published. According to Ohasi et al. (37), the potent proinflammatory response by the human heat shock protein 60 was absent in macrophages from mutant C3H/HeJ TLR4 mice, whereas they responded to CpG DNA. Macrophages from normal mice had a proinflammatory response to the heat shock protein 60, suggesting activation of TLR4. Besides this protein, other endogenous mediators are candidates as activators of TLR. The fact that recruitment of TLR2, TLR1, and TLR6 was induced by phagosomes containing IgG-coated erythrocytes suggests the role of immunocomplexes as activators of TLR (32). Inasmuch as mice with deleted MyD88 gene have apparently normal embryonic development, it is unlikely that a TLR response controls embryonic development.
CLINICAL IMPLICATIONS
There are remarkable differences in the tolerance of mucosal surfaces for microbes. The number of bacteria in the gastrointestinal tract normally exceeds the number of somatic cells, but the nonpathogenic microbes do not induce the cytokine response. Other mucous membranes (peripheral airways, alveoli, and urinary tract) are generally sterile, despite their intermittent exposure to microbes. The differences in responsiveness remain largely unexplained. Apart from the cytokine cascade apparently controlled by TLRs, other microbial clearance mechanisms include specific antibodies, mucociliary escalator, phagocytosis, intracellular killing by phagocytes, and production of proteins involved in host defense (collectins, defensins, lysozyme, lactoferrin, transferrin, and others). MBL, SP-A, and SP-D of the collectin family bind to CD14, LPS, and other toxins (38, 39). However, a collectin may promote microbial clearance without activation of the NF-κB–based inflammatory cascade (40).
Most TLRs are expressed in several tissues, although often at very low levels. Besides the contribution to innate immunity, the potential roles of TLRs in signaling tissue destruction, chronic organ injury, differentiation, and neoplastic disease remain to be studied. A defect or abnormality in one component of the innate immune system may predispose to disease first in the presence of exceptional stress (41).
The innate immune system has critical roles in host defense during periods of transition, particularly during the early development of clonal immunity, when maternal immunoglobulins are present at low levels. In intrauterine infections, the unprepared host is affected by microbes or proinflammatory cytokines, with serious consequences (fetal death owing to shock, susceptibility to long-term organ injury) (42). The expression levels of the germ line–encoded components of host defense depend on the stage of differentiation. This deficient or abnormal responsiveness of the innate immune system in immature lung is in line with the variably low expression levels of SP-A (43), SP-D (44), TLR2, and TLR4 (45). The responsiveness of lung to cytokines and microbial components is complex and age-dependent, suggesting effective developmental regulation of the responsiveness of innate immunity (46, 47). Small premature infants with immature lungs may exhibit bronchopulmonary dysplasia, characterized by a lack of growth and alveolarization and deficient gas exchange (48). Increased chemokines and proinflammatory cytokines and low antiinflammatory IL-10 (49) and SP-A (43) associate with chronic lung disease, possibly as a sign of maladaptation of the immune system.
Susceptibility to recurrent infections is influenced by heredity and may, to some extent, involve microbial-binding proteins. A lack of TLR4 expression predisposes mice to Gram-negative bacterial infection (50), and an absence of the scavenger receptor, to Staphylococcus aureus infection (51). Furthermore, defects in complement enhance bacterial infections both in mice (52) and in humans (53). A lack of SP-A in mice enhances infections originating from the respiratory tract, most notably Group B Streptococcus and Pseudomonas(41). In humans, deficient expression of MBL because of allelic variation has been associated with meningococcal disease (54, 55) and severe cystic fibrosis complicated with Pseudomonas aeruginosa infection (56). A particular SP-A haplotype, on the other hand, associates with susceptibility to recurrent respiratory infections in infants (57). Allelic variation of human TLR4 has been shown to influence airway responses in LPS challenge (26).
Whether any defect in the pattern recognition proteins influences the incidence or severity of allergies and autoimmune diseases remains to be studied (58, 59).
CONCLUSIONS
Allelic variation in the genes involved in innate immunity may account for individual differences in the responses to corresponding pathogens. For example, a single nucleotide polymorphism in the TLRs could explain some of the clinical heterogeneity of the responses to an infectious challenge (26). Another example of how allelic gene variation modifies the balance between host and pathogen is the MBL gene that correlates with differences in life expectancy among patients with identical cystic fibrosis genotypes (56). The role of different TLRs in such diseases as cystic fibrosis as well as the efforts to ascertain whether polymorphisms in TLRs play a role in modifying the innate responses in this and other childhood diseases will inevitably attract a great deal of attention. Deficient or altered proteins involved in microbial binding or subsequent signaling potentially lead to an inappropriate acquired immune response (54, 55, 59). The role of innate immunity is critical during early childhood, when the clonal development of antibodies and other host defenses is inadequate. It would seem that premature infants are particularly vulnerable to subtle dysregulation of innate immunity. Understanding the role of each TLR is the first step toward determining the effects that variations in TLR expression and primary sequence have on modifying the host response to an infectious challenge. This knowledge may help us develop novel preventive and therapeutic strategies.
Abbreviations
- CpG:
-
cytosine phosphate-guanosine
- IL-1RI:
-
IL-1 type I receptor
- IRAK:
-
IL-1 receptor-associated kinase
- LPS:
-
lipopolysaccharide
- LRR:
-
leucine-rich repeat (segment of extracellular part of TLR)
- MBL:
-
mannose-binding lectin
- NF:
-
nuclear transcription factor
- SP:
-
surfactant protein
- TIR:
-
domain, Toll-IL-1 receptor domain (cytoplasmic part of TLR, IL-1 and IL-18)
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor alpha
References
Medzhitov R, Janeway CA Jr 1997 Innate immunity: the virtues of a nonclonal system of recognition. Cell 91: 295–298
Hoffmann JA, Kafatos FC, Janeway CA Jr, Ezekowitz RA 1999 Phylogenetic perspectives in innate immunity. Science 284: 1313–1318
Stahl PD, Ezekowitz RA 1998 The mannose receptor is a pattern recognition receptor involved in host defense. Curr Opin Immunol 10: 50–55
Anderson KV, Jurgens G, Nusslein-Volhard C 1985 Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell 42: 779–789
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA 1996 The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86: 973–983
Medzhitov R, Preston-Hulburt P, Janeway CA Jr 1997 A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388: 394–397
Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF 1998 A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA 95: 588–593
Chaudhary PM, Ferguson C, Nguyen V, Nguyen O, Masa FM, Eby M, Jasmin A, Trask BJ, Hood L, Nelson PS 1998 Cloning and characterization of two Toll/interleukin-1 receptor-like genes TIL3 and TIL4: evidence for a multi-gene receptor family in humans. Blood 91: 4020–4027
Takeuchi O, Kawai T, Sanjo H, Copeland NG, Gilbert DJ, Jenkins NA, Takeda K, Akira S 1999 TLR6: a novel member of an expanding Toll-like receptor family. Gene 231: 59–65
Du X, Poltorak A, Wei Y, Beutler B 2000 Three novel mammalian toll-like receptors: gene structure, expression, and evolution. Eur Cytokine Netw 11: 362–371
Imler J-L, Hoffmann JA 2000 Signalling mechanisms in the antimicrobial host defense of Drosophila. Curr Opin Microbiol 3: 16–22
Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogava T, Takeda K, Akira S 1999 Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as Lps gene product. J Immunol 162: 3749–3752
Poltorak A, Ricciardi-Castagnoli P, Citterio S, Beutler B 2000 Physical contact between lipopolysaccharide and Toll-like receptor 4 revealed by genetic complementation. Proc Natl Acad Sci USA 97: 2163–2167
Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M 1999 MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189: 1777–1782
Muzio M, Ni J, Feng P, Dixit VM 1997 IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278: 1612–1615
Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A 1998 The human Toll signaling pathway: divergence of nuclear factor kappa B and JNK/SAPK activation upstream of tumor factor receptor-associated factor 6 (TRAF6). J Exp Med 187: 2097–2101
DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M 1997 A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature 388: 548–554
Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M 1997 The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell 91: 243–252
Rothwarf DM, Zandi E, Natoli G, Karin M 1998 IKK-γ is an essential regulatory subunit of the IκB kinase complex. Nature 395: 297–300
Sulzer BM 1968 Genetic control of leukocyte responses to endotoxin. Nature 219: 1253–1254
McAdam KWPJ, Ryan JL 1978 C57BL/10/CR mice: nonresponders to activation by the lipid A moiety of bacterial lipopolysaccharide. J Immunol 120: 249–253
Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B 1998 Defective LPS signalling in C3H/HeJ and C57/BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085–2088
Du X, Poltorak A, Silva M, Beutler B 1999 Mutational analysis of Tlr4-mediated LPS signal transduction in macrophages. Blood Cells Mol Dis 25: 328–338
Beutler B 2000 Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 12: 20–26
Kuhns DB, Long PD, Gallin JI 1997 Endotoxin and IL-1 hyporesponsiveness in a patient with recurrent bacterial infections. J Immunol 158: 3959–3964
Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA 2000 TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet 25: 187–191
Yang R-B, Mark MR, Gurney AL, Godowski PJ 1999 Signaling events induced by lipopolysaccharide-activated Toll-like receptor 2. J Immunol 163: 639–643
Kirsching CJ, Wesche H, Merrill AT, Rothe M 1998 Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J Exp Med 188: 2091–2097
Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S 1999 Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11: 443–451
Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A 1999 The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401: 811–814
Schwandler R, Dziarski R, Wesche H, Rothe M, Kirsching CJ 1999 Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J Biol Chem 274: 17406–17409
Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A 2000 The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc Natl Acad Sci USA 97: 13766–13771
Lipford GB, Heeg K, Wagner H 1998 Bacterial DNA as immune cell activator. Trends Microbiol 6: 496–500
Hemmi H, Takeuchi O, Kawai T, Kalsho M, Hoshino K, Wagner H, Takeda K, Akira S 2000 A Toll-like receptor recognizes bacterial DNA. Nature 408: 740–745
Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, Kelly RA 1999 Toll (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest 104: 271–280
Muzio M, Polentarutti B, Bosisio D, Prahladan MK, Mantovani A 2000 Toll-like receptors:: a growing family of immune receptors that are differentially expressed and regulated by different leukocytes. J Leukoc Biol 67: 450–456
Ohashi K, Burkart V, Flohe S, Kolb H 2000 Cutting edge: heat shock protein 60 is a putative endogenous ligand of the Toll-like receptor-4 complex. J Immunol 164: 558–561
Crouch EC 1998 Collectins and pulmonary host defense. Am J Respir Cell Mol Biol 19: 177–201
Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y 2000 Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem 275: 22442–22451
Wright JR, Zlogar DF, Taylor JC, Zlogar TM, Restrepo CI 1999 Effects of endotoxin on surfactant protein A and D stimulation of NO production by alveolar macrophages. Am J Physiol 276: L650–L658
Korfhagen TR, LeVine AM, Whitsett JA 1998 Surfactant protein A (SP-A) gene targeted mice. Biochim Biophys Acta 1408: 296–302
Hallman M 1999 Cytokine, pulmonary surfactant and consequences of intrauterine infection. Biol Neonate 76: 2–9
Hallman M, Arjomaa P, Mizumoto M, Akino T 1988 Surfactant proteins in the diagnosis of fetal lung maturity. I. Predictive accuracy of the 35 kD protein, the lecithin/sphingomyelin ratio, and phosphatidylglycerol. Am J Obstet Gynecol 158: 531–535
Miyamura K, Malhotra R, Hoppe HJ, Reid KB, Phizackerley PJ, Macpherson P, Lopez Bernal A 1994 Surfactant proteins A (SP-A) and D (SP-D): levels in human amniotic fluid and localization in the fetal membranes. Biochim Biophys Acta 1210: 303–307
Pelkonen K, Glumoff V, Hallman M 2001 Ontogeny of Toll-like receptors Tlr2 and Tlr4 in mouse. Pediatr Res 49: 81–83
Bry K, Lappalainen U, Hallman M 1997 Intraamniotic interleukin-1 accelerates surfactant protein synthesis in fetal rabbits and improves lung stability after premature birth. J Clin Invest 99: 2992–2999
Glumoff V, Väyrynen O, Kangas T, Hallman M 2000 Degree of lung maturity determines the direction of the interleukin-1-induced effect on the expression of surfactant proteins. Am J Respir Cell Mol Biol 22: 280–288
Jobe A 1999 The new BPD: an arrest of lung development. Pediatr Res 46: 641–643
Speer CP, Groneck P 1998 Oxygen radicals, cytokines, adhesion molecules and lung injury in neonates. Semin Neonatol 3( suppl): 219–228
O'Brien AD, Rosenstreich DL, Scher I, Cambell GH, Macdermott RP, Formal SB 1980 Genetic control of susceptibility to Salmonella typhimurium in mice: role of the LPS gene. J Immunol 124: 20–24
Thomas CA, Li Y, Kodama T, Suzuki H, Silverstein SC, Khoury JE 2000 Protection from lethal gram-positive infection by macrophage scavenger receptor-dependent phagocytosis. J Exp Med 191: 147–155
Fisher MB, Prodeus AP, Nicholson-Weller PA, Ma M, Murrow J, Reid RR, Warren HB, Lage AL, Moore FD Jr, Carroll MC 1997 Increased susceptibility to endotoxin shock in complement C3- and C4-deficient mice is corrected by C1 inhibitor replacement. J Immunol 159: 976–982
Buckley RH 1992 Immunodeficiency in diseases. JAMA 268: 2797–2806
Hibberd ML, Sumiya M, Summerfield JA, Booy R, Levin M, and Meningococcal Research Group 1999 Association of variants of the gene for mannose-binding lectin with susceptibility to meningococcal disease. Lancet 353: 1049–1053
Bax WA, Cluysenaer OJ, Bartelink AK, Aerts PC, Ezekowitz RA, van Dijk H 1999 Association of familial deficiency of mannose-binding lectin and meningococcal disease. Lancet 354: 1094–1095
Garred P, Pressler T, Madsen HO, Frederiksen B, Svejgaard A, Hoiby N, Schwartz M, Koch C 1999 Association of mannose-binding lectin gene hetero-geneity with severity of lung disease and survival in cystic fibrosis. J Clin Invest 104: 431–437
Rämet M, Löfgren J, Alho O-P, Hallman M 2001 Surfactant protein-A gene locus associated with recurrent otitis media. J Pediatr 138: 266–268
Garred P, Madsen HO, Marquart H, Hansen TM, Sorensen SF, Petersen J, Volck B, Svejgaard A, Graudal NA, Rudd PM, Dwek RA, Sim RB, Andersen V 2000 Two edged role of mannose binding lectin in rheumatoid arthritis: a cross sectional study. J Rheumatol 27: 26–34
Eggleton P, Reid KBM 1999 Lung surfactant proteins involved in innate immunity. Curr Opin Immunol 11: 28–33
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the Academy of Finland [M.H., M.R.], the Biocenter Oulu, Sigrid Jusélius Foundation [M.H.], and the Foundation for Pediatric Research [M.H., M.R.].
Rights and permissions
About this article
Cite this article
Hallman, M., Rämet, M. & Ezekowitz, R. Toll-like Receptors as Sensors of Pathogens. Pediatr Res 50, 315–321 (2001). https://doi.org/10.1203/00006450-200109000-00004
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200109000-00004
This article is cited by
-
Sialylated human milk oligosaccharides prevent intestinal inflammation by inhibiting toll like receptor 4/NLRP3 inflammasome pathway in necrotizing enterocolitis rats
Nutrition & Metabolism (2021)
-
Toll-like receptor-mediated innate immunity against herpesviridae infection: a current perspective on viral infection signaling pathways
Virology Journal (2020)
-
LncRNA HOTAIR regulates lipopolysaccharide-induced cytokine expression and inflammatory response in macrophages
Scientific Reports (2018)
-
Immunomodulatory effects of lactoferrin
Acta Pharmacologica Sinica (2014)
-
Anti-inflammatory Properties of Orange Juice: Possible Favorable Molecular and Metabolic Effects
Plant Foods for Human Nutrition (2013)
