
Abstract
We present a premature newborn of 32 wk of gestation with a congenital malignant extrarenal rhabdoid tumor (MERT) on the right shoulder with generalized metastases. Standard histologic, immunohistochemical, molecular and cytogenetic methods were used in the evaluation of diagnostic material. Biopsy of a skin lesion showed the histologic features of a malignant rhabdoid tumor. Cytogenetic analysis of the tumor cells revealed an inv(11)(p13p15) and additionally, an increased expression of myf-3 (myogenic determination factor, MyoD1) and PAX3 was detected. These results suggest an origin of the neoplasm derived from a pluripotent cell with the potential of myogenic differentiation. Tumor suppressor genes located on chromosome 11p13 and 11p15 may play an important role for malignant rhabdoid tumor development and progression.
Similar content being viewed by others
Main
Malignant extrarenal rhabdoid tumor (MERT) is a rare highly malignant tumor of unknown origin, mainly found in small children and with a mostly fatal clinical outcome. We present a case of a premature female newborn of 32 wk of gestation with a large s.c. tumor on the right shoulder and disseminated metastases already detected by sonography at the 30th week of gestation. Histopathology, molecular- and cytogenetic examinations suggest a pluripotent cell with the possibility of myogenic differentiation.
MERT shares similar histologic, immunohistochemical, ultrastructural, and clinical features with malignant renal rhabdoid tumor (MRRT) (1–4). Many extrarenal locations have been described including the CNS, liver, soft tissue, and skin (5–10). The median age at presentation is 10 to 13 mo. Histologically, the neoplasm consists of a monotonous population of large polygonal tumor cells with eccentric nuclei and prominent nucleoli. Inclusions of eosinophilic hyaline aggregates displacing the nucleus are commonly seen. Electron microscopy demonstrates specific large cytoplasmic whorls of intermediate filaments and no signs of differentiation. Immunohistochemically the cells are uniformly vimentin positive, all other immunostainings have a variable reactivity suggesting tissue heterogeneity and various cell differentiation (1, 11–14).
A 32-year-old gravida-5 para-3 had an uneventful pregnancy until 31 wk of gestation when she was admitted to the hospital for premature labor. A routine scan at 20 wk of gestation was normal. The mother had previously delivered three healthy boys, one pregnancy ended in an early spontaneous abortion.
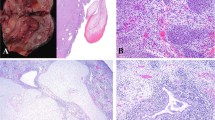
An abdominal ultrasound at 31 wk of gestation revealed a 5.6 × 8.8 cm cystic-solid tumor at the right shoulder (Fig. 1) and a sacral tumor measuring 1.5 × 1.5 cm. Due to a symptomatic polyhydramnion 1000 mL of the amniotic fluid was drained. Subsequently i.v. tocolysis and steroids for pulmonary maturation were administered. Daily ultrasound examinations demonstrated no further increase of the amniotic fluid index. Because variable decelerations occurred at 32 wk of gestation reflecting fetal distress, cesarean section was performed. The birth weight of the female newborn was 1820 g. The Apgar score was 5/8/9, umbilical vein pH 7.14. Initially, the general condition could be stabilized after primary resuscitation, however, respiratory distress occurred and at the age of two hours mechanical ventilation was necessary.

Prenatal ultrasound at the 31st week of gestation demonstrating a cystic-solid tumor (5.6 cm × 8.8 cm) on the right shoulder (arrows).
At the right shoulder a s.c. tumor of 9 × 5 × 3 cm was seen involving the neck and the upper thorax discoloring the skin (Fig. 2). Generalized multiple skin metastases were present. The child did not show dysmorphic signs. Chest x-ray showed the soft tissue mass located at the right shoulder with tiny amorphous calcifications, whereas skeletal lesions were not seen. Abdominal ultrasound revealed hepatomegaly with several lesions suspicious of metastases. The spleen, kidneys, and adrenal glands were morphologically normal. A peripheral blood smear revealed an aggregation of undifferentiated cells, supposed to be tumor cells. The bone marrow aspirate showed a diffuse infiltration of the undifferentiated cells. Initial Hb was 123 g/L and no tumor lysis syndrome was detected in the laboratory results. Alpha-fetoprotein and beta-choriogonadotropin were in the upper normal range (174′710 μg/L and 44 IU/L, respectively), urine catecholamines were negative. Arterial hypotension and renal insufficiency with oliguria occurred in the follow-up. Uric acid increased to 480 mM with no electrolyte abnormalities. Blood transfusions and prophylaxis for tumor lysis syndrome were given. A malignant rhabdoid tumor was diagnosed based on a skin biopsy. No specific therapy for the tumor was given. The infant died 6 days after birth of congestive heart failure. A postmortem examination was performed.

Large s.c. tumor at the right shoulder of approximately 9 × 5 × 3 cm.
METHODS
Histopathology and Immunohistochemistry.
For light microscopy, tissue samples were fixed in formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin, Giemsa and periodic acid-Schiff (PAS). Immunostaining was performed with the following antibodies: vimentin, Lu-5, epithelial membrane antigen, neuron specific enolase, Myf-3, smooth muscle actin (SMA), desmin, myoglobin, actin (HHF 35), S-100 Protein, leu 7 (CD 47), CD99 (HBA71-B), CD34, CD20, factor VIII, β-HCG (beta-choriogonadotropin), alpha-fetoprotein, and carcino-embryonal antigen (CEA).
For electron microscopy tissue was fixed in glutaraldehyde, embedded in Epon and contrasted with uranyl acetate and lead citrate. Ultrathin sections were studied with a electron microscope Zeiss EM 900.
Cytogenetics.
Cytogenetic analyses were performed on bone marrow (BM) and peripheral blood (PB) samples. The cells were suspended in RPMI 1640 with Glutamax 1 (GIBCO, Paisley, Scotland) supplemented with 20% fetal bovine serum and 1% penicillin/streptomycin. The BM cells were in culture for 20 to 48 h and the PB cells for 72 h. The PB cultures were set up with different cell concentrations and stimulated with phytohemagglutinin (PHA). All cultures were harvested according to recognized cytogenetic methods with the resulting fixed cells being trypsin banded and Giemsa stained. Analysis was made according to ISCN 1995 (15).
RNA Analysis.
To further characterize this tumor, expression of several markers were investigated by a reverse transcriptase PCR assay (RT-PCR). These markers included the t(1;13) and t(2;13) translocation products PAX3/FKHR and PAX7/FKHR that are associated with alveolar rhabdomyosarcoma as well as the native PAX3 and PAX7 gene products. PAX genes are transcription factors, which act as essential control genes during the establishment of embryonic cell lineages, particularly myogenesis and neurogenesis (16). In addition, the expression of the myogenic human myf3 gene was examined. Myf3 is a specific marker for all cells of myogenic origin (17).
RNA isolated from different rhabdomyosarcoma cell lines was included as a positive control. For RNA isolation the BM cells were recovered by a Ficoll-Hypaque density gradient centrifugation. Total RNA was extracted using TRIzol (GIBCO-BRL) according to the manufacturer's instructions and treated with DNase I (PharMingen). The RT-PCR was performed using the Access RT-PCR System Kit (Promega) in a 25 μL reaction with 100 ng total RNA. As a positive control, RNA from the rhabdomyosarcoma cell lines RD (FKHR, PAX3, PAX7 and Myf-3) or Rh30 (PAX3PAX7/FKHR) was used. All reactions started with reverse transcription at 48°C for 45 min, continued by an initial denaturation step (94°C, 2 min.), followed by 40 cycles at 94°C for 30 s, 60°C or 68°C for 1 min and 68°C for 1 min with a final extension at 68°C for 7 min. The 68°C annealing temperature was used only for the Myf-3 amplification. The FKHR, PAX3, and PAX7 products were generated in 0.5 mM MgSO4 and the PAX3PAX7/FKHR, and Myf-3 in 1.0 mM MgSO4. The following primers were used to amplify a 324-bp long fragment of FKHR (FKHRF3 5′-AACTGTGATCCAGGGCTGTC-3′ and FKHR-F5 5′GCAGATCTACGAGTGGATGG-3′), a 349-bp fragment of PAX3 (PAX3–5′ 5′-GCACTGTACACCAAAGCACG-3′ and PAX3-REV 5′-TAGGTGGGTGGACAGTAGGA-3′), a 414-bp fragment of PAX7 (PAX7–3 5′GGCGTAAGCAGGCAGGAG-3′ and PAX7–4 5′-GCAGCGGG- GAGATGGAGA-3′), a 868-bp or 862-bp fragment for the translocations PAX3/FKHR or PAX7/FKHR (PAX3PAX7 5′-CCAAACACAGCATCGACG-3′, this primer anneals to both PAX3 and PAX7, and FKHR-F3 5′-AACTGTGATCCAGGGCTGTC-3′), and finally a 159-bp fragment of Myf-3 (Myf-3-FOR 5′-CTGTGGGCCTGCAAGGCGTGCAAG-3′, and MYF3-REV 5′-CACCTTGGGCAACCGCTGGTTTGG-3′). One third of the RT-PCR products was separated by electrophoresis on a 2% agarose gel.
Informed consent was received from the parents, and the study was approved by the ad hoc ethical committee of the University Children's Hospital of Basel.
RESULTS
Histology and Immunohistochemistry.
The histologic sections of all involved tumor sites including the small renal lesion revealed the same aspect of homogenous undifferentiated tumor cells infiltrating the tissue. Lungs showed extended infiltration of the pulmonary vessels and lymphatic system. The monomorphic tumor cells were round to polygonal with eosinophilic cytoplasm, large vesicular nuclei and prominent nucleoli with homogenous, eosinophilic globular cytoplasmic inclusions (Fig. 3). Mitoses were frequently seen. Electron microscopy revealed intermediate filaments often arranged in whorls near the nucleus. No signs of differentiation were seen (Fig. 4). Immunohistochemically the cells and particularly the intracellular inclusions were positive for vimentin, Lu-5, epithelial membrane antigen and neuron specific enolase. Myf-3 showed a weak positivity. The following immunostainings were negative: smooth muscle actin (SMA), desmin, myoglobin, actin (HHF 35), S-100 Protein, leu 7 (CD 47), CD99 (HBA71-B), CD34, CD20, factor VIII, β-HCG (beta-choriogonadotropin), alpha-fetoprotein and carcino-embryonal antigen (CEA).

Malignant extrarenal rhabdoid tumor from the right shoulder showing polygonal cells with typical eosinophilic globular cytoplasmic inclusions (H&E) (arrows).

Electron microscopy of malignant extrarenal rhabdoid tumor cells revealing paranuclear whorls of intermediate filaments (Zeiss EM 900).
Autopsy.
The large gray-white, homogenous tumor infiltrating the right shoulder presented with a diameter of 7.5 cm. Multiple generalized metastases with diameters of up to 3 cm were seen in the skin, liver, lungs, both adrenal glands, retroperitoneal lymph nodes and soft tissue, mesenterium, thymus, pancreas, dura, and skeleton including ribs and vertebrae. In the kidneys a single tiny lesion of 6 mm in diameter was seen. No tumor tissue was found in the brain. In addition, placental examination showed neither macroscopic nor microscopic evidence for metastasis.
Cytogenetics.
Analysis of the bone marrow revealed an abnormal karyotype of 46,XX,inv(11) (p13p15)[8])/46,XX[90]). The percentage of abnormal cells was found to be higher in the 48 h rather than in the shorter term cultures. The inversion could also be found in one PB culture.
Molecular biology.
The patient's tumor cells showed a clearly detectable expression of myf3, at a level similar to the rhabdomyosarcoma control, as well as low level expression of PAX3 (Fig. 5). In contrast, neither a specific translocation product nor any PAX7 message was detected.

RT-PCR analysis of the bone marrow metastasis RNA (lane 1, 3, 5, 7, and 9). As control for the RT-PCR reactions control RNA from rhabdomyosarcoma cell lines were used (lanes 2, 4, 6, 8, and 10). To assure the integrity of the total RNAs FKHR was amplified by RT-PCR as a control. The specificity of the PAX3 RT-PCR products was confirmed by direct sequencing.
DISCUSSION
Etiology, pathogenesis and clinical features of malignant renal and extrarenal rhabdoid tumor have been debated since the first description in 1978 by Beckwith and Palmer (18). The tumor may represent an evolution of various neoplasms which progress to a more aggressive biologic behavior as the occurrence of rhabdoid cells had been demonstrated in different tumor types (1, 19–24). To our knowledge we describe for the first time a malignant extrarenal rhabdoid tumor of a premature infant at 32 wk of gestation. The aggressive clinical behavior and the typical cytological and immunohistological findings lead to the diagnosis. The large solid tumor mass on the right shoulder is suggestive of the primary tumor site, a renal origin seems unlikely. Despite the generalized metastases the placenta was free of tumor suggesting a possible maternal mechanism of tumor cell recognition and destruction.
Chromosomal abnormalities of rhabdoid tumors have been found often to involve chromosomal regions 22q11 and 11p15.5 (25–30). Aberrations of the long arm of chromosome 22 were first demonstrated in a MERT of the CNS (31). Recently, Schofield et al. (32) showed loss of heterozygosity (LOH) on chromosome 22 in 24 of 30 malignant renal rhabdoid tumors and five of them had LOH at 11p15.5. We describe a MERT of a premature newborn with an inv(11)(p13p15). Both affected regions on chromosome 11 are known to be locations of tumor suppressor genes. The Wilms' tumor gene (WT) 1 is located at 11p13. In a recent study, 3 MRRT were positive for the WT1 gene product (33). In another case of MRRT a del(11)(p13) was described (34). Garvin et al. (35) showed that the G401 cell line from a supposed Wilms' tumor actually derived from a MRRT. This cell line was used for studies demonstrating the presence of a tumor suppressor gene on chromosome 11 (36). The WT2 locus maps to chromosome 11p15.5 and is affected in patients with the Beckwith-Wiedemann syndrome (BWS), who have a an increased risk of developing Wilms' tumor (37). Newsham et al. (38) reported a MERT cell line (TM 87–16) with t(11;22)(p15.5;q11.23). Lee et al. (39) demonstrated that KVLQT1, a previously identified gene located on chromosome 11p15.5, is disrupted by chromosomal rearrangement in this MERT cell line (TM 87–16) and in BWS patients. Consistent allelic loss of chromosomal region 11p15 has been also found in embryonal rhabdomyosarcoma (40). Our findings suggest that the disruption of the regions 11p13 and 11p15 with a possible loss of tumor suppressor gene or genes may play a role in rhabdoid tumor genesis or progression. WT1 and WT2 could be candidate genes, but further studies are required to support this idea.
Recently the hSNF5/INI1 gene has been described as a new tumor suppressor gene located on chromosome 22q11.2, and has shown to be mutated in MRT (41). The gene encodes a member of the ATP-dependent chromatin remodeling SWi/SNF multiprotein complexes, which are thought to facilitate the access of transcription factors to DNA. Sévenet et al. screened 229 tumor samples of various origins and found the mutation in most MRT (53/72), however the mutation was also detected in choroid plexus carcinomas (4/6), central primitive neuroectodermal tumors (cPNET) (2/17) and medulloblastomas (5/36) (42). In addition they analyzed constitutional DNA from affected members in cancer-prone families. Their data indicate, that constitutional mutations of the hSNF5/INI1 gene exist and could lead to a hereditary syndrome predisposing to MRT and some tumors of the CNS (43).
The demonstration of PAX3 expression in our case is a new finding in a MERT. PAX3 is transiently expressed in early skeletal muscle progenitor cells and in the developing nervous system. The gene has a possible role of stimulating growth, inhibiting apoptosis or inhibiting premature myogenic differentiation and participates in critical developmental processes. PAX7 and both fusion products (PAX3/FKHR, PAX7/FKHR) typical for alveolar rhabdomyosarcomas, were not expressed (40).
Myf3 is a specific human myogenic determination factor regulating muscle differentiation (44). The expression of the gene in our case would indicate a myogenic origin of the tumor cells. Gessler et al. (45) localized the gene for myf3 on chromosome 11p14. However, they found no association with the WAGR locus (Wilms' tumor, aniridia, genitourinary anomalies, and mental retardation) at 11p13, the location of WT1, or the region for the BWS locus at 11p15, the location of WT2. Dias et al. (46) examined the immunohistochemical expression of MyoD1 in a variety of pediatric solid tumors. A MRRT showed weak, nonspecific MyoD1 staining. Two malignant rhabdoid cell lines failed to demonstrate MyoD1 immunofluorescence staining and did not express MyoD1 transcripts whereas the rhabdomyosarcomas expressed MyoD1. Some Wilms' tumors demonstrated focal staining for MyoD1 antiserum. The presence of heterotopic tissue may reflect primitive developmental potential of metanephric blastema.
The expression of myf3 and PAX3 in a MERT in early fetal life together with all classical clinical, histologic, and immunohistochemical findings suggest an origin of the neoplasm derived from a pluripotent cell with the potential of myogenic differentiation. An undifferentiated rhabdomyosarcoma with rhabdoid features cannot be excluded but seems to be unlikely (47, 48). An overexpression of PAX3 could exaggerate its normal role, affects the cellular activities and may lead to an oncogenic effect.
Abbreviations
- DNA/RNA:
-
desoxyribonucleid/ribonucleid acid
- MRT:
-
malignant rhabdoid tumor
- MERT/MRRT:
-
malignant extrarenal/renal rhabdoid tumor
- WT1/WT2:
-
Wilms' tumor suppressor gene 1 and 2
References
Wick MR, Ritter JH, Dehner LP 1995 Malignant rhabdoid tumor: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol 12: 233–248
Kodet R, Newton WA, Sachs N, Hamoudi AB, Raney BR, Asmari L, Gehan EA 1991 Rhabdoid tumors of soft tissues. Hum Pathol 22: 674–684
Gururangan S, Bowman LC, Parham DM, Wilimas J, Rao B, Pratt CB, Douglass EC 1993 Primary extracranial rhabdoid tumors. Cancer 71: 2653–2659
Schmidt D, Leuschner I, Harms D, Sprenger E, Schäfer H-J 1989 Malignant rhabdoid tumor. Path Res Pract 184: 202–210
Weeks DA, Malott RL, Zuppan CW, Liwnicz BH, Beckwith JB 1994 Primitive cerebral tumor with rhabdoid features: a case of phenotypic rhabdoid tumor of the central nervous system. Ultrastruct Pathol 18: 23–28
Behring B, Brück W, Goebel HH, Behnke J, Pekrum A, Christen HJ, Kretzschmar HA 1996 Immunohistochemistry of primary central nervous system malignant rhabdoid tumors: report of five cases and review of the literature. Acta Neuropathol 91: 578–586
Bhattacharjee M, Hicks J, Dauser R, Strother D, Chintagumpala M, Horowitz M, Cooley L, Vogel H 1997 Primary malignant rhabdoid tumor of the central nervous system. Ultrastruct Pathol 21: 361–368
Hunt SJ, Anderson WD 1990 Malignant rhabdoid tumor of the liver. Am J Clin Pathol 94: 645–648
Tsokos M, Kouraklis G, Chandra RS, Bhagavan BS, Triche TJ 1989 Malignant rhabdoid tumor of the kidney and soft tissues. Arch Pathol Lab Med 113: 115–120
Perez-Atayde AR, Newbury R, Fletcher JA, Barnhill R, Gellis S 1994 Congenital “neurovascular hamartoma” of the skin. Am J Surg Pathol 18: 1030–1038
Weeks DA, Beckwith JB, Mierau GW, Luckey DW 1989 Rhabdoid tumor of kidney. Am J Surg Pathol 13: 439–458
Tsuneyoshi M, Daimaru Y, Hashimoto H, Enjoji M 1987 The existence of rhabdoid cells in specified soft tissue sarcomas. Virchows Arch A 411: 509–514
Sotelo-Avila C, Gonzales-Crussi F, deMello D, Vogler C, Gooch WM, Gale G, Pena R 1986 Renal and extrarenal rhabdoid tumors in children: a clinicopathologic study of 14 patients. Semin Diagn Pathol 3: 151–163
Kaiserling E, Ruck P, Handgretinger R, Leipoldt M, Hipfel R 1996 Immunohistochemical and cytogenetic findings in malignant rhabdoid tumor. Gen Diagn Pathol 141: 327–337
Mitelman F 1995 An International System of Human Cytogenetic Nomenclature. S. Karger, Basel, pp 62
Schäfer BW 1998 Emerging roles for PAX transcription factors in cancer biology. Gen Physiol Biophys 17: 211–224
Goulding MD, Chalepakis G, Deutsch U, Erselius JR, Gruss P 1991 Pax-3, a novel murine DNA binding protein expressed during early neurogenesis. EMBO 10: 1135–1147
Beckwith JB, Palmer NF 1978 Histopathology and prognosis of Wilms' tumor. Cancer 41: 1937–1948
Weeks DA, Beckwith JB, Mierau GW 1989 Rhabdoid tumor. Arch Pathol Lab Med 113: 113–114
Weeks DA, Beckwith JB, Mierau GW, Zuppan CW 1991 Renal neoplasms mimicking rhabdoid tumor of kidney. Am J Surg Pathol 15: 1042–1054
Tsuneyoshi M, Daimaru Y, Hashimoto H, Enjoji M 1987 The existence of rhabdoid cells in specified soft tissue sarcomas. Virchows Arch A 411: 509–514
Kodet R, Newton WA, Hamoudi AB, Asmar L 1991 Rhabdomyosarcomas with intermediate-filament inclusions and features of rhabdoid tumors. Am J Surg Pathol 15: 257–267
Parham DM, Weeks DA, Beckwith JB 1994 The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. Am J Surg Pathol 18: 1010–1029
Perlman EJ, Ali SZ, Robinson R, Lindato R, Griffin C 1998 Infantile extrarenal rhabdoid tumor. Pediatr Dev Pathol 1: 149–152
Handgretinger R, Kimming A, Koscielnak E, Schmidt D, Rudolph G, Wolburg H, Paulus W, Schilbach-Stueckle K, Ottenlinger C, Menrad A, Sproll M, Bruchelt G, Dopfer R, Treuner J, Niethammer D 1990 Establishment and characterization of a cell line (Wa-2) derived from an extrarenal rhabdoid tumor. Cancer Res 50: 2177–2182
Sait SN, Nowak NJ, Singh-Kahlon P, Weksberg R, Squire J, Shows TB, Higgins MJ 1994 Localization of Beckwith-Wiedemann and rhabdoid tumor chromosome rearrangements to a defined interval in chromosome band 11p15. Genes Chromosom Cancer 11: 97–105
Shashi V, Lovell MA, von Kap-herr C, Waldron P, Golden WL 1994 Malignant Rhabdoid tumor of the kidney: Involvement of Chromosome 22. Genes Chromosom Cancer 10: 49–54
Hirose M, Yamada T, Abe T, Hirose T, Shimizu E, Yamamoto Y, Kagami S, Takano S, Yamaguchi T, Kuroda Y 1996 Establishment and characterization of two cultured cell lines derived from malignant rhabdoid tumors of the kidney. Int J Cancer 67: 218–223
Hirose M, Yamada T, Toyosaka A, Hirose T, Kagami S, Abe T, Kuroda Y 1996 Rhabdoid tumor of the kidneys: a report of two cases with respective tumor markers and specific chromosomal abnormality del(11p13). Med Pediatr Oncol 27: 174–178
Douglass EC, Valentine M, Rowe ST, Parham DM, Wilimas JA, Sanders JM, Houghton PJ 1990 Malignant rhabdoid tumor: a highly malignant childhood tumor with minimal karyotypic changes. Genes Chromosom Cancer 2: 210–216
Biegel JA, Allen CS, Kawasaki K, Shimizu N, Budarf ML, Bell CJ 1996 Narrowing the critical region for a rhabdoid tumor locus. Genes Chromosom Cancer 16: 94–105
Schofield DE, Beckwith JB, Sklar J 1996 Loss of heterozygosity at chromosome regions 22q11–12 and 11p15. Genes Chromosom Cancer 15: 10–17
Ramani P, Cowell JK 1996 The expression pattern of Wilms' tumour gene (WT1) product in normal tissues and pediatric renal tumors. J Pathol 179: 162–168
Kent AL, Mahoney DH, Gresik MV, Steuber CP, Fernbach DJ 1987 Malignant rhabdoid tumor of the extremity. Cancer 60: 1056–1059
Garvin AJ, Re GG, Tarnowski BI, Hazen-Martin DJ, Sens DA 1993 The G401 cell line, utilized for studies of chromosomal changes in Wilms' tumor, is derived from a rhabdoid tumor of the kidney. Am J Pathol 142: 375–380
Dowdy SF, Fasching CL, Araujo D, Lai K-M, Livanos E, Weissman BE, Stanbridge EJ 1991 Suppression of tumorigenicity in Wilms' tumor by the p15. Science 254: 293–295
Besnard-Guérin C, Newsham I, Windqvist R, Cavenee WK 1996 A common region of loss of heterozygosity in Wilms' tumor and embryonal rhabdomyosarcoma distal to D11S988 locus on chromosome 11p15. Hum Genet 97: 163–170
Newsham I, Daub D, Besnard-Guérin C, Cavenee W 1994 Molecular sublocalization and characterization of the 11;22 translocation breakpoint in a malignant rhabdoid tumor. Genomics 19: 433–440
Lee MP, Hu R-J, Johnson LA, Feinberg AP 1997 Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nature Genet 15: 181–185
Barr FG 1997 Molecular genetics and pathogenesis of rhabdomyosarcoma. J Pediatr Hematol Oncol 19: 483–491
Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O 1998 Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394: 203–206
Sévenet N, Lellouch-Tubiana A, Schofield D, Hoang-Xuan K, Gessler M, Birnbaum D, Jeanpierre C, Jouvet A, Delattre O 1999 Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet 8: 2359–2368
Sévenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O 1999 Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65: 1342–1348
Schäfer BW, Blakely BT, Darlington GJ, Blau HM 1990 Effect of cell history on response to helix-loop-helix family of myogenic regulators. Nature 344: 454–458
Gessler M, Hameister H, Henry I, Junien C, Braun T, Arnold HH 1990 The human MyoD1 (Myf3) gene maps on the short arm of chromosome 11 but is not associated with the WAGR locus or the region for the Beckwith-Wiedemann syndrome. Hum Genet 86: 135–138
Dias P, Parham DM, Shapiro DN, Webber BL, Houghton PJ 1990 Myogenic regulatory protein (MyoD1) expression in childhood solid tumors. Am J Pathol 137: 1283–1291
Pautard B, Canarelli JP, Gontier MF, Risbourg R, Piussan C, Flamant F 1990 Rhabdomyosarcome embryonnaire de découverte anténatale. Arch Fr Pediatr 47: 433–435
Lobe TE, Wiener ES, Hays DM, Lawrence WH, Andrassy RJ, Johnston J, Wharam Webber B, Rgab A 1994 Neonatal rhabdomyosarcoma: the IRS experience. J Pediatr Surg 29: 1167–1170
Acknowledgements
The authors thank Paul Imbach for reviewing the manuscript and for helpful discussions.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Staehelin, F., Bissig, H., Hösli, I. et al. Inv(11)(p13p15) and Myf-3(MyoD1) in a Malignant Extrarenal Rhabdoid Tumor of a Premature Newborn. Pediatr Res 48, 463–467 (2000). https://doi.org/10.1203/00006450-200010000-00008
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200010000-00008
