
Abstract
We have studied activation of circulating polymorphonuclear leukocytes(PMN) in plasma of preterm infants with severe idiopathic respiratory distress syndrome (IRDS group, n = 15) and without IRDS (reference group,n = 15) during the first 5 postnatal days. We have observed lower median PMN counts in the IRDS group than in the reference group from d 2 (1.4× 109/L versus 4.8 × 109/L in the reference group, p < 0.001) to d 4 to 6 (1.6 × 109/Lversus 4.0 × 109/L, p < 0.01). Lower PMN counts in the IRDS infants were accompanied by lower median plasma elastase-α1-proteinase inhibitor (PI) concentrations (53.6 ng/mLversus 128.0 ng/mL in the reference group on d 2, p < 0.05). Simultaneously, median elastase-α1-PI/PMN ratios of these infants were significantly higher (40.8 ng/106 PMN versus 21.8 ng/106 PMN on d 2, p < 0.05), indicating activation of circulating PMN. Activation of circulating PMN in the IRDS group is associated with platelet-activating factor (PAF) release and complement activation from within 6 to 12 h of birth but not with release of tumor necrosis factor-α. PAF release was represented by significantly reduced inhibiting capacity (58% of normal human plasma, p < 0.01) and complement activation by higher median plasma C3a des-Arg concentrations (1680 ng/mL versus 325 ng/mL in the reference group, p < 0.001). We conclude that circulating PMN are activated in preterm infants with severe IRDS, which might be caused by systemic PAF release and complement activation. This activation process may play a role in the pathogenesis of the IRDS by influx of activated PMN into the lungs.
Similar content being viewed by others


Main
An early severe inflammatory reaction has been found in the lungs of preterm infants with the IRDS and appears to be involved in the development of BPD. This inflammatory reaction is characterized by accumulation and activation of leukocytes(1–5) and local release of inflammatory mediators(1, 2, 4–10). Activated leukocytes are predominantly PMN and alveolar macrophages(1–5). Release of elastase by activated PMN in the presence of low levels of the main proteinase inhibitorα1-PI has been associated with lung connective tissue destruction and the development of BPD(1, 2, 4, 5, 7). These destructive effects of elastase are aggravated by pneumonia or hyperoxic exposure(11–13). Activated alveolar macrophages release more TNF-α in infants with IRDS, who develop BPD, compared with those with an uncomplicated course(9, 10). Furthermore, inflammatory mediators such as PAF(6) leukotriene B4(4, 5), IL-1β(5), IL-8(4), and complement component C5-derived anaphylatoxin(C5a)(4, 8) have been found in tracheal aspirates of infants at risk for BPD. These mediators likely contribute to respiratory insufficiency by several mechanisms including attraction and activation of PMN and breakdown of pulmonary vascular endothelium with subsequent protein leakage into the small airways(4–6, 14, 15). This pulmonary protein-rich edema is thought to play an important role in the pathogenesis of BPD(16).
Similar to infants with IRDS, accumulation of activated PMN in the lungs is found in children and adults with severe clinical disorders that result in the ARDS. This influx of PMN into the lungs is accompanied by a decrease of the circulating PMN count and massive injury of lung tissue due to local release of oxygen radicals and proteolytic enzymes (e.g. elastase)(17–19). Several inflammatory mediators are thought to play a role in PMN activation and accumulation in the lungs of ARDS patients(17, 19, 20). However, some of these mediators do not always act exclusively in the airways. Increase of the TNF-α plasma concentration and systemic activation of complement have been described in ARDS patients(19–22). Furthermore, systemically administered PAF mediates lung tissue injury in animal experimental studies(23).
We questioned whether accumulation of PMN in the lungs of preterm infants with IRDS is accompanied by activation of circulating PMN with subsequent systemic release of elastase. Circulating inflammatory mediators including TNF-α, activated complement compounds, and PAF might be involved in this activation process. Therefore, this study was undertaken to obtain more insight in the process of PMN activation in preterm infants with IRDS. We measured 1) the total leukocyte count, 2) the PMN and monocyte count, 3) the plasma concentration of elastase-α1-PI complex (indicator of PMN activation),4) TNF-α (indicator of monocyte activation), 5) PAF-IC in plasma (indicator of PAF release), and 6) the plasma C3a des-Arg concentration (indicator of complement activation) simultaneously during the first 5 d of life in preterm infants with severe IRDS and in preterm infants without respiratory problems.
METHODS
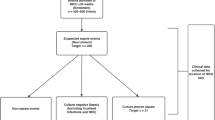
Patients. Thirty preterm infants consecutively admitted to the neonatal intensive care unit of the Beatrix Childrens Hospital, University Hospital Groningen, were included in this study. These infants fitted the following criteria for study enrolment: 1) no maternal infection, amnionitis, or prolonged rupture of membranes (>24 h before birth);2) gestational age between 27 and 33 wk; 3) birth weight appropriate for gestational age; and 4) no major congenital malformations and/or evidence of infection. Fifteen infants showed clinical and radiologic signs of severe IRDS (IRDS group). Severe IRDS was defined as oxygen requirement of more than 30% for adequate oxygenation, artificial ventilation dependency because of respiratory insufficiency, and grade 3 or 4 abnormalities on a chest x-ray according to the Giedion score(24). Fifteen preterm infants without IRDS or other significant problems served as a reference group. The study was approved by the hospital ethical committee, and informed consent was obtained from the parents of all infants.
There were no differences between the IRDS and the reference infants for prenatal factors that could influence activation of circulating PMN, release of TNF-α PAF, or activation of complement (Table 1). The mothers of four infants in the IRDS group and of four infants in the reference group showed HELLP and/or toxicosis before delivery. Pregnant women, who develop HELLP syndrome show complement activation at delivery(25). However, complement components do not cross the placenta(26). About half of the newborn infants whose mothers have HELLP and/or toxicosis during pregnancy are known to have diminished PMN counts at birth(27). However, PMN counts in the four reference infants could not be distinguished from the other reference infants, whereas PMN counts in the four IRDS infants were lower than those of the four reference infants. Dexamethasone was given more than 2 d before delivery to the mothers of five infants in both the IRDS and the reference groups. Dexamethasone does not clearly affect plasma complement activation, but decreases leukocyte activation and release of bioactive mediators(28) and increases activity of PAF-acetylhydrolase, the natural occurring inhibitor of PAF(29). However, elastase-α1-PI and TNF-α plasma concentrations and the PAF-IC in the five IRDS and the five reference infants did not differ from the other infants in each group.
Postnatal characteristics of the IRDS and the reference group are presented in Table 1. All IRDS infants had significantly lower 1- and 5-min Apgar scores and significantly lower arterial umbilical pH values than the reference infants. Despite of these signs of birth asphyxia the IRDS infants showed no other organ failure but respiratory insufficiency from birth. All IRDS infants in this study required endotracheal intubation at birth and were artificially ventilated throughout the study period.
Patient management. Infants admitted with hypoxia and respiratory insufficiency were artificially ventilated with supplemental oxygen to maintain arterial Po2 between 7.5 and 10.0 kPa, and Pco2 between 5.5 and 6.5 kPa. When infants showed clinical signs of respiratory distress, required more than 30% oxygen for adequate oxygenation and artificial ventilation because of respiratory insufficiency, they were eligible for the Dutch Multicenter study “Nedsurf” for surfactant replacement therapy (Alvofact, Boehringer Ingelheim, Germany). In the Nedsurf study, IRDS infants are prospectively classified according to the roentgenologic severity of the disease using the Giedion score(24). Infants showing Giedion 1 or 2 abnormalities on a chest x-ray were considered to have moderate IRDS, whereas infants with a Gideon 3 or 4 chest x-ray were considered to have severe IRDS. In our study all infants of the IRDS group had severe IRDS. According to the criteria of the Nedsurf study, 11 of the 15 infants of the IRDS group received surfactant(100 mg/kg body weight endotracheally). The remaining four infants were not given surfactant because of bloody tracheal secretions.
In all study infants superficial cultures were obtained immediately after admission and on d 4. None of these infants showed colonization with pathogenic bacteria, especially those, that are known to be predominant causes of early onset septicemia in our neonatal intensive care unit. In 12 IRDS infants, cultures of blood, tracheal effluent, and urine were taken because of a decrease of the leukocyte count. The blood, tracheal effluent, and urine cultures remained negative in 10 of these 12 infants. These 10 infants were included in the present report and did not show other clinical signs of an infection but respiratory failure, which was thought to be caused by IRDS. Two other infants were excluded from the study. One of these infants showed a pneumonia/sepsis with group B streptococci, whereas another infant showed anEscherichia coli sepsis.
According to the local protocol, infants of less than 34 wk of gestational age with respiratory insufficiency requiring continuous positive airway pressure or artificial ventilation were screened with echocardiography for a patent ductus arteriosus on the 2nd to 4th d of life. When the ductus arteriosus was open, these infants were treated with indomethacin (Indocid, Merck Sharpe & Dohme, Haarlem, The Netherlands) i.v. Seven infants of the IRDS group were treated for an open ductus arteriosus.
Infants received transfusions of cryoglobulin-poor plasma or packed red blood cells with cryoglobulin-poor plasma for replacement of blood taken for routine laboratory investigations. The transfusion volume for each infant in the study did not exceed 10% of the calculated blood volume in any 24-h period. Furthermore, cryoglobulin-poor plasma is stored frozen and contains only native inactive plasma proteins, whereas packed red blood cells contain only 10 to 20% plasma, including plasma proteins, which are slightly activated by the processing procedure(30). Therefore, replacement transfusions are not considered to influence the findings of this study in the IRDS infants nor the reference infants.
Insertion or removal of a venous or arterial umbilical catheter or a radial artery catheter was approved by the attending neonatologist. The patency of arterial umbilical catheters and arterial radial catheters was maintained by continuous infusion of a 0.9% NaCl solution containing 3 U/mL heparin at a rate of 0.5 to 0.7 mL/h.
Heparin is known to inhibit the complement system(31). We have tried to minimize effect of heparin on complement activation in the samples of our IRDS infants. In these infants, blood samples were obtained from the venous umbilical catheter, which was not heparinized nor flushed with heparin before collecting the actual blood sample to be used for analysis. Although we did not measure the heparin concentration in the blood sample that was analyzed, this should be very low. It has been demonstrated that complement inhibition occurs especially with heparin concentrations similar to those needed for anticoagulation. However, despite heparinization, complement activation has still been demonstrated in humans and animals undergoing extracorporeal life support(32, 33). Furthermore, it has been shown that the heparin infusion that we used does not influence clotting in preterm infants(34). We, therefore, do not consider heparin as used in our infants of great influence on the results of our study. All infants of the IRDS group had a venous umbilical catheter, whereas 10 of them also had an arterial umbilical catheter (n = 7) or an arterial radial catheter(n = 3). The infants of the reference group did not receive an indwelling catheter.
Study protocol. Blood samples were taken from the venous umbilical catheter (IRDS infants) or were obtained by venipuncture (reference infants) at 6 to 12 h from birth (1st d of life), and on the 2nd, 3rd, and 4th to 6th d of life. The first blood sample was taken before treatment with surfactant, indomethacin, or blood products was performed. Each blood sample was taken during routine blood sampling and used for the determination of the total leukocyte count, the PMN and monocyte count, the plasma concentration of elastase-α1-PI and TNF-α, the plasma PAF-IC, and the activation of complement (C3a des-Arg).
At each sampling 0.3 mL blood was anticoagulated with EDTA (0.01 M) for determination of the total leukocyte count and differential cell counts. Another 0.5 mL of blood was anticoagulated with citrate (0.3%). This citrated blood sample was centrifuged at room temperature immediately after collection. After centrifugation the plasma sample was immediately stored at -80°C. Because blood sampling had to be as limited as possible and only one blood sample per study infant could be centrifuged and stored immediately after collection, we decided to determine the concentration of elastase-α1-PI, C3a des-Arg, and PAF-IC in this sample. With this technique even better results were obtained with regard to elastase-α1-PI concentrations and PAF-IC than with centrifuging EDTA samples after cell counting was performed. The latter, however, would be a better alternative for determination of C3a des-Arg regarding in vitro activation of complement, which was now limited by rapid processing to storage of the citrated samples. The elastase-α1-PI/PMN ratio was calculated to exclude the influence of the circulating PMN count on the systemic release of elastase. Also the TNF-α/monocyte ratio was calculated to exclude the influence of the circulating monocyte count on the systemic release of TNF-α. Both ratios are expressed in nanograms/106 PMN or monocytes
Assays. The total nucleated cell count was determined using a cell counter (Hemolog, Coulter Electronics, UK). The total leukocyte count was determined after correction of the total nucleated cell count for the presence of nucleated red cells (erythroblasts). The leukocytes were differentiated by morphologic classification of 100 cells in blood films, which were stained by the May-Grünwald-Giemsa method.
Plasma elastase-α1-PI concentrations were quantitated by an ELISA (Merck, Darmstadt, Germany). TNF-α was determined with a RIA(TNF-α IRMA, Medgenix, Brussels, Belgium). The C3a des-Arg plasma concentration was determined by RIA (Upjohn Co., Kalamazoo, MI).
For the determination of the PAF-IC, platelets were isolated from citrated platelet-rich plasma of healthy adult volunteers by filtration through Sepharose CL-2B (Pharmacia Biotech Inc., Stockholm, Sweden). These isolated platelets were resuspended in PBS to a final platelet concentration of 50× 109/L. A 700-μL aliquot of this platelet suspension was dispensed into a cuvette, incubated at 37°C, and stirred at 900 rpm with a metallic rod within an aggregometer (Chronolog Corp., Havertown, PA) which recorded the light transmission pattern of the platelet suspension. Then a 50-μL aliquot of plasma of the study infants or normal human adults was then added to the cuvette after 5 min followed by 5 μL of the PAF C16 suspension (10 mg/mL, Cayman Chemical, Ann Arbor, MI). Platelet aggregation after addition of PAF was allowed to continue until maximal increase in light transmission. The percentage of platelet aggregation after addition of PAF is inversely correlated with the PAF-IC of plasma. The PAF-IC is expressed as the percentage of maximal PAF inhibition in normal human plasma.
Statistical analysis. Data are presented as mean ± SD or as median with 25th and 75th percentiles as appropriate. The χ2 test with Yates's correction for continuity was used for comparison of nominal data between the IRDS and the reference group. Comparison of gestational age, birth weight, 1- and 5-min Apgar scores and arterial umbilical pH values between the two groups was carried out using the unpaired t test.
For total leukocyte counts and TNF-α, two-way analysis of variance for repeated measures was used to test the effects of time and group-time interactions. In addition, an unpaired t test was performed to compare the values of these parameters between the two groups on d 1, 2, 3, and 4 to 6. A paired t test was used to compare total leukocyte count and TNF-α on d 2, 3, and 4 to 6 with the values of these parameters on d 1.
For leukocyte differential cell counts, plasma elastase-α1-PI and C3a des-Arg concentrations, plasma elastase-α1-PI/PMN and TNF-α/monocyte ratio, and the PAF-IC, means of five observations were calculated for each individual infant and compared between the IRDS and the reference group, thus testing intergroup difference for this parameter. The Mann-Whitney U test and the Wilcoxon signed rank test were used to determine specific differences between and within the two groups for the PMN and monocyte count, the plasma elastase-α1-PI and C3a des-Arg concentration, the plasma elastase-α1-PI/PMN and TNF-α/monocyte ratio, and the PAF-IC.
The significance level was adjusted for multiple comparisons by means of the Bonferroni correction as appropriate. Statistical significance was assumed when the p value was less than 0.05.
RESULTS
Ventilatory support. The mean Fio2 value in the IRDS infants decreased significantly from 0.72 ± 0.14 on d 1 to 0.46± 0.25 on d 5 (p < 0.05). Peak inspiratory pressure values did not change significantly between d 1 and 5 (24 ± 3 cm H2Oversus 23 ± 8 cm H2O) in these infants. Two reference infants showed apnea and bradycardia and required nasal continuous positive airway pressure without supplemental oxygen for 1 and 4 d, respectively. One reference infant showed signs of a wet lung shortly after birth and needed endotracheal intubation and artificial ventilation during the 1st d of life with maximal Fio2 and peak inspiratory pressure values of 0.27 and 18 cm H2O, respectively. The remaining 12 reference infants showed no respiratory problems.
Total leukocyte count. The total leukocyte count (mean ± SD) of the IRDS infants was significantly lower than that of the reference infants (p = 0.001, analysis of variance for repeated measures). The difference between both study groups was significant at each sampling point(Fig. 1). In the IRDS group, the total leukocyte count decreased from 5.4 ± 1.9 × 109/L on d 1 to 3.5 ± 2.0 × 109/L on d 2 (p < 0.05 versus d 1) and increased afterward to 5.1 ± 2.2 × 109/L on d 4 to 6. In the reference group, the total leukocyte count did not change significantly between d 1 and d 4 to 6.

Total leukocyte count in preterm infants with(open circles) and without IRDS (closed circles) during the first 5 d of life. Data are expressed as mean ± SD. *p< 0.05, **p < 0.01, ***p < 0.001 for IRDS group compared with reference group.
PMN count. The median PMN count (25th to 75th percentile) of the IRDS group was significantly lower than that of the reference group from the 2nd d of life (Fig. 2). The median PMN count did not change significantly in both study groups throughout the study period.

PMN count in preterm infants with (open boxes) and without IRDS (gray boxes) during the first 5 d of life. Data are presented as box graphs showing the median values(horizontal plane line) ranges of 50% around the median value(boxes), and the 10th and 90th percentile (error bars).**p < 0.01, ***p < 0.001 for IRDS group compared with reference group.
Elastase -α1-PI complex and elastase -α1-PI/PMN ratio. From d 2, the median plasma elastase-α1-PI complex concentration (25th to 75th percentile) of the IRDS infants was significantly lower than that of the reference infants (Fig. 3). The median plasma elastase-α1-PI complex concentration did not change significantly during the first 5 d of life in both groups.

Plasma concentrations of elastase-α1-PI complex in preterm infants with (open boxes) and without IRDS(gray boxes) during the first 5 d of life. Data are presented as box graphs showing the median values (horizontal plane line), ranges of 50% around the median value (boxes), and the 10th and 90th percentile (error bars). *p < 0.05 for IRDS group compared with reference group.
From d 2, the median elastase-α1-PI/PMN ratio (25th to 75th percentile) of the IRDS group was significantly higher than that of the reference group (Fig. 4). The median elastase-α1-PI/PMN ratio did not show significantly changes within each study group throughout the study period.

Elastase-α1-PI/PMN ratio in preterm infants with (open boxes) and without IRDS (gray boxes) during the first 5 d of life. Data are presented as box graphs showing the median values (horizontal plane line), ranges of 50% around the median value (boxes), and the 10th and 90th percentile (error bars). *p < 0.05 for IRDS group compared with reference group.
Monocyte count. The median monocyte count (25th to 75th percentile) of the IRDS group was significantly lower than that of the reference group from the 3rd day of life (0.3 (0.08-0.4) × 109/Lversus 0.8 (0.5-0.9) × 109/L on d 3, p < 0.01; 0.4 (0.2-0.9) × 109/L versus 1.1 (1.0-1.6)× 109/L on d 4 to 6, p < 0.05). The median monocyte count within each study group did not change during the first 5 d of life.
TNF -α and TNF -α/ monocyte ratio. The plasma concentration of TNF-α (mean ± SD) of the IRDS group did not differ from that of the reference group (p = 0.244, analysis of variance for repeated measures). The TNF-α concentration did not change significantly in the IRDS group (8.0 ± 7.5 ng/mL on d 1; 13.7 ± 11.5 ng/mL on d 4 to 6) and in the reference group (13.2 ± 9.1 ng/mL on d 1; 17.1 ± 9.2 ng/mL on d 4 to 6) throughout the study period. During the first 5 d of life the median TNF-α/monocyte ratio of both groups was not different and did not change significantly.
PAF-IC. The median PAF-IC (25th to 75th percentile) of the IRDS infants was significantly lower than that of the reference group on d 1, 2, and 4 to 6 (Fig. 5). The median PAF-IC of both groups did not change significantly during the first 5 d of life.

Plasma PAF-IC in preterm infants with (open boxes) and without IRDS (gray boxes) during the first 5 d of life. Data are presented as box graphs showing the median values(horizontal plane line), ranges of 50% around the median value(boxes), and the 10th and 90th percentile (error bars).*p < 0.05, **p < 0.01 for IRDS group compared with reference group.
C3a des-Arg. During the first 5 d of life the median C3a des-Arg plasma concentration (25th to 75th percentile) of the IRDS infants was higher than that of the reference infants (Fig. 6). The median C3a des-Arg plasma concentration decreased in the IRDS group without being significant. The median C3a des-Arg plasma concentration in the reference group remained low during the first 5 d of life and did not change significantly.

Plasma concentrations of C3a des-Arg in preterm infants with (open boxes) and without IRDS (gray boxes) during the first 5 d of life. Data are presented as box graphs showing the median values(horizontal plane line), ranges of 50% around the median value(boxes), and the 10th and 90th percentile (error bars).**p < 0.01, ***p < 0.001 for IRDS group compared with reference group.
DISCUSSION
In this study, artificially ventilated preterm infants with respiratory failure from birth and clinical and radiologic signs of severe IRDS (IRDS group) showed a lower total leukocyte count than preterm infants without respiratory problems (reference group). This lower total leukocyte count was due to a lower PMN and monocyte count. In the IRDS infants the lower PMN count was accompanied by a lower plasma elastase-α1-PI concentration but a higher elastase-α1-PI/PMN ratio than in the reference infants. The latter suggests systemic activation of circulating PMN in the IRDS infants. Simultaneously, indications were obtained for systemic PAF release and complement activation but not for systemic TNF-α release in these infants.
From the 2nd d of life, the PMN count was significantly lower in the IRDS group than in the reference group. This lower PMN count probably reflects influx of PMN into the lungs, which has been reported in preterm infants with severe IRDS(1–5). Simultaneously, we observed lower elastase-α1-PI concentrations in the IRDS infants than in the reference infants in accordance with findings of Ogden et al.(1). The plasma concentrations of elastase-α1-PI of the reference infants did not differ from those of cord blood of healthy term newborn infants(35). The low plasma concentrations of elastase-α1-PI in the IRDS infants might be explained by the lower circulating PMN count that will reduce the absolute amount of elastase to be released. To exclude this influence of the circulating PMN count on the plasma elastase-α1-PI concentration, we have calculated the plasma elastase-α1-PI/PMN ratio. This ratio was significantly higher in the IRDS group than in the reference group from the 3rd d of life, indicating increased release of elastase by circulating PMN in the IRDS infants. This increased release may be caused by circulating inflammatory mediators such as TNF-α, PAF, and complement split products(19–23).
Our data do not indicate systemic release of TNF-α in the IRDS infants. Actually, plasma TNF-α concentrations of our IRDS infants were slightly but not significantly lower than those of our reference infants, similarly to the findings of Murch et al.(9). TNF-α is mainly produced and released by activated monocytes and macrophages(36). Circulating monocytes are known to produce less TNF-α than stimulated alveolar macrophages(37). From d 3, we have observed a significantly lower monocyte count in the IRDS infants than in the reference infants, which might explain the lower TNF-α concentrations in the IRDS infants. In addition, we could not observe increased TNF-α release by circulating monocytes by using the TNF-α/monocyte ratio, which confirms the limited TNF-α producing capacity of circulating monocytes. Thus, release of TNF-α seems to be a localized phenomenon as demonstrated by high TNF-α concentrations found in bronchopulmonary secretions of infants with IRDS, who subsequently develop BPD(9, 10).
Throughout the study period the inhibition of PAF by plasma of the IRDS infants was lower than by plasma of the reference infants. PAF inhibition is represented by PAF-acetylhydrolase, which is consumed by complexing PAF(38). PAF inhibiting capacity is decreased in newborn preterm infants due to preexisting low plasma activity of PAF-acetylhydrolase(39). However, PAF inhibition was lower in the IRDS group than in the reference group, being significant on d 1, 2, and 4 to 6. Therefore, the lower inhibition of PAF in plasma of our IRDS infants can be considered as an indication of PAF-acetylhydrolase consumption due to increased PAF release in the circulation. Release of PAF in blood has been demonstrated in preterm ventilated infants during the 1st wk of life(40) and confirms our findings in the IRDS infants. Systemic release of PAF is likely to contribute to (further) respiratory insufficiency in the IRDS infants by systemic activation of PMN and complement, thus mediating increased pulmonary vascular permeability with formation of protein-rich edema(15, 16, 41).
In the IRDS group, activation of the complement system was represented by higher plasma concentrations of C3a des-Arg, the inactive split product of C3. Increased C3a des-Arg plasma concentrations are described in preterm and term infants with respiratory insufficiency, who had birth asphyxia or circulatory insufficiency(42), and in term neonates with severe respiratory failure requiring extracorporeal life support(35). Recently, Groneck et al.(4, 8) have found that the complement anaphylatoxin C5a is present in plasma and tracheobronchial aspirate fluid of preterm infants with IRDS at risk for chronic lung disease. These reports support our findings that the complement system is activated in newborn infants with severe respiratory distress. C3a and C5a are known to increase vascular permeability(43) and might contribute to pulmonary edema formation in IRDS infants. Furthermore, C5a contributes to PMN chemotaxis, aggregation, and local sequestration in the lungs(20).
We found higher C3a des-Arg plasma concentrations in both our reference and IRDS infants than was demonstrated by others(44). This might be explained by differences in methods. We sampled blood with citrate instead of EDTA. Citrate probably allows more in vitro activation of the complement system than EDTA, but this was limited by rapid processing of the samples to storage at -80°C. Most importantly, we used a commercially available RIA. In this assay polyclonal antibodies to C3a also measures determinants present on the native C3 molecule. Burger et al.(45) used a highly sensitive C3a assay that measures C3a without being affected by the presence of C3.
The C3a des-Arg values found in our IRDS infants were higher than those found in our reference infants. We did not find evidence of infections in these infants as demonstrated by negative cultures of blood, tracheal effluent, and urine. The low TNF-α plasma concentrations further confirm the absence of infections in our IRDS infants because very high TNF-α plasma concentrations have been demonstrated in newborn infants with sepsis(46). However, apart from infections, several explanations can be proposed for the high C3a des-Arg values in our IRDS infants. First, all of these infants showed low Apgar scores and low arterial umbilical pH values at birth, indicating perinatal asphyxia (hypoxemia and acidosis). Hypoxemia and tissue hypoperfusion are accompanied by release of activated complement components(47), which might explain the high C3a des-Arg plasma concentrations shortly after birth. Second, all IRDS infants required artificial ventilation with high peak inspiratory pressures and high Fio2 values throughout the study period. Tissue trauma is accompanied by local complement activation(48). The aforementioned C5a in tracheobronchial aspirate fluid of preterm infants with IRDS at risk for BPD(4, 8) probably leaks into the circulation. Third, in ventilated infants with severe IRDS, increased plasma factor XII and kallikrein activity has been described(49, 50). Both activated factor XII and kallikrein are able to induce complement activation(51). Finally, low plasma concentrations of C1 esterase inhibitor has been described in preterm infants(52). C1 esterase inhibitor is one of the main inhibitors of the complement system. Release of activated complement products out of the lungs together with activated factor XII and kallikrein and low plasma concentrations of C1 esterase inhibitor might explain the high C3a des-Arg plasma concentrations in our IRDS infants throughout the study period.
The cause of the lower PAF-IC values in our IRDS infants also is not yet clarified. Similar to activation of complement, activation of PAF may be caused by hypoxemia due to birth asphyxia(53) and tissue injury due to artificial ventilation(6). The production and release of inflammatory mediators such as PAF and activated complement factors may become self-perpetuating. Systemic release of PAF causes vasoconstriction, thus contributing to tissue ischemia in several organs(41) and subsequent activation of complement(47). In turn, complement activation causes production and release of more PAF by activating PMN(54).
In summary, we have found that activation of circulating PMN represented by increased elastase-α1-PI/PMN ratios occurs simultaneously with a low circulating PMN count in preterm infants with severe IRDS during the first five days of life. We suggest that these activated PMN have more localized effects because no other organ failure but respiratory insufficiency occurred in the IRDS infants. Furthermore, we have found indications for systemic PAF release and complement activation. Hypoxemia, tissue ischemia due to perinatal asphyxia, and lung tissue injury due to artificial ventilation may all have induced PAF release and complement activation. We did not find evidence of any infection in our IRDS infants. In preterm infants, systemic release of PAF and activated complement factors may play a role in the pathogenesis of severe IRDS by chemotaxis and sequestration of activated PMN into the lungs. However, further studies in preterm infants with mild, moderate and severe IRDS are required to determine whether systemic PAF release and complement activation in these infants is related to IRDS severity and local inflammation in the lung.
Abbreviations
- IRDS:
-
idiopathic respiratory distress syndrome
- BPD:
-
bronchopulmonary dysplasia
- ARDS:
-
adult respiratory distress syndrome
- PMN:
-
polymorphonuclear leukocytes
- α1-PI:
-
α1-proteinase inhibitor
- TNF-α:
-
tumor necrosis factor α
- PAF:
-
platelet-activating factor
- IC:
-
inhibiting capacity of plasma
- Fio2:
-
fractional concentration of inspired O2
- HELLP:
-
hemolysis, elevated liver enzymes, and low platelet count syndrome
References
Ogden BE, Murphy SA, Saunders GC, Pathak D, Johnson JD 1984 Neonatal lung neutrophils and elastase/proteinase inhibitor imbalance. Am Rev Respir Dis 130: 817–821.
Merritt TA, Cochrane CG, Holcomb K, Bohl B, Hallman M, Strayer D, Edwards DK 1983 Elastase and α-1-proteinase inhibitor activity in tracheal aspirates during respiratory distress syndrome.. J Clin Invest 72: 656–666.
Jackson JC, Chi EY, Wilson CB, Truog WE, Teh EC, Hodson WA 1987 Sequence of inflammatory cell migration into the lung during recovery from hyaline membrane disease in premature newborn monkeys. Am Rev Respir Dis 135: 937–940.
Groneck P, Götze-Speer B, Oppermann M, Eiffert H, Speer CP 1994 Association of pulmonary inflammation and increased microvascular permeability during the development of bronchopulmonary dysplasia: a sequential analysis of inflammatory mediators in respiratory fluids of high-risk preterm neonates. Pediatrics 93: 712–718.
Groneck P, Reuss D, Götze-Speer B, Speer CP 1993 Effects of dexamethasone on chemotactic activity and inflammatory mediators in tracheobronchial aspirates of preterm infants at risk for chronic lung disease. J Pediatr 122: 938–944.
Stenmark KR, Eyzaguirre M, Westcott JY, Henson PM, Murphy RC 1987 Potential role of eicosanoids and PAF in the pathophysiology of broncho-pulmonary dysplasia. Am Rev Respir Dis 136: 770–772.
Speer CP, Reuss D, Harms K, Herting E, Gefeller O 1993 Neutrophil elastase and acute pulmonary damage in neonates with severe respiratory distress syndrome. Pediatrics 91: 794–799.
Groneck P, Oppermann M, Speer CP 1993 Levels of complement anaphylatoxin C5a in pulmonary effluent fluid of infants at risk for chronic lung disease and effects of dexamethasone treatment. Pediatr Res 34: 586–590.
Murch SH, MacDonald TT, Wood CBS, Costeloe KL 1992 Tumor necrosis factor in the bronchoalveolar secretions of infants with the respiratory distress syndrome and the effect of dexamethasone treatment. Thorax 47: 44–47.
Murch SH, MacDonald T, Costeloe K 1994 Macrophage-associated chemokines and cytokines in neonatal respiratory distress syndrome. Early Hum Dev 38: 60
Bruce M, Wedig KE, Jentoft N, Martin K, Cheng PW, Boat TF, Fanaroff AA 1985 Altered urinary excretion of elastin cross-links in premature infants who develop bronchopulmonary dysplasia. Am Rev Respir Dis 131: 568–572.
Bruce MC, Schuyler M, Martin RJ, Starcher BC, Tomashefski JF, Wedig KE 1992 Risk factors for the degradation of lung elastic fibers in the ventilated neonate. Am Rev Respir Dis 146: 204–212.
Walti H, Tordet C, Gerbaut L, Sauger P, Moriette G, Relier JP 1989 Persistent elastase/proteinase inhibitor imbalance during prolonged ventilation of infants with bronchopulmonary dysplasia: evidence for the role of nosocomial infections. Pediatr Res 26: 351–355.
Vercelotti GM, Yin HQ, Gustafson KS, Nelson PO, Jacob HS 1988 Platelet-activating factor primes neutrophil responses to agonists: role in promoting neutrophil-mediated endothelial damage. Blood 137: 1364–1370.
Hamaski Y, Mojarad M, Saga T, Tai H, Said SI 1984 Platelet-activating factor raises airway and vascular pressures and induces edema in lungs perfused with platelet-free solution. Am Rev Respir Dis 129: 742–746.
O'Brodovich HM, Coates G 1988 Pulmonary edema in respiratory distress syndrome and bronchopulmonary dysplasia. In: Merritt TA, Northway WH, Boynton BR (eds) Bronchopulmonary Dysplasia. Blackwell Scientific Publications, Boston, 143–159.
Roall JA, Levin DL 1987 Adult respiratory distress syndrome in pediatric patients: I. Clinical aspects, pathophysiology, and mechanisms of lung injury. J Pediatr 112: 169–180.
Sarnaik AP, Lieh-Lai M 1994 Adult respiratory distress syndrome in children. Pediatr Clin North Am 41: 337–364.
Suter PM, Suter S, Girardin E, Roux-Lombard P, Grau GE, Dayer JM 1992 High bronchoalveolar levels of tumor necrosis factor and its inhibitors, interleukin-1, interferon, and elastase, in patients with adult respiratory distress syndrome after trauma, shock, or sepsis. Am Rev Respir Dis 145: 1016–1022.
Robbins RA, Russ WD, Rasmussen JK, Clayton MM 1987 Activation of the complement system in the adult respiratory distress syndrome. Am Rev Respir Dis 135: 651–658.
Marks JD, Marks CB, Luce JM, Montgomery AB, Turner J, Metz CA, Murray JF 1990 Plasma tumor necrosis factor in patients with septic shock. Mortality rate, incidence of adult respiratory distress syndrome and effects of methyl-prednisolone administration. Am Rev Respir Dis 141: 94–97.
Weinberg PF, Mathay MA, Webster RO, Roskos KV, Goldstein IM, Murray JF 1984 Biologically active products of complement and acute lung injury in patient with the sepsis syndrome. Am Rev Respir Dis 130: 791–796.
Chang SW, Feddersen CO, Henson PM, Voelkel NF 1987 Platelet-activating factor mediates hemodynamic changes and lung injury in endotoxin-treated rats. J Clin Invest 79: 1498–1509.
Giedion A, Haefliger H, Dangel P 1973 Acute pulmonary x-ray changes in hyaline membrane disease treated with artificial ventilation and positive end expiratory pressure. Pediatr Radiol 1: 145–152.
Haeger M, Unander M, Bengtsson A 1991 Complement activation in relation to development of preeclampsia. Obstet Gynecol 78: 46–49.
Propp RA, Alper CA 1968 C3 synthesis in the human fetus and lack of transplacental passage. Science 162: 672–673.
Koenig JM, Christensen RD 1989 Incidence, neutrophil kinetics, and natural history of neonatal neutropenia associated with maternal hypertension. N Engl J Med 321: 557–562.
Jansen NJG, van Oeveren W, van Vliet M, Stoutenbeek CP, Eysman L, Wildevuur Ch RH 1991 The role of different types of corticosteroids in the inflammatory mediators in cardiopulmonary bypass. Eur J Cardiothorac Surg 5: 211–217.
Furukawa M, Lee EL, Johnston JM 1993 Platelet-activating factor-induced ischemic bowel necrosis: the effect of platelet-activating factor acetylhydrolase. Pediatr Res 34: 237–241.
Gu YJ, Obster R, de Haan J, Gallandat Huet RCG, van Oeveren W 1992 Biocompatibility of leukocyte removal filters during bedside leukocyte filtration of red cell concentrates. Transfus Sci 13: 467–472.
Kazatchkine MD, Fearon DT, Metcalfe DD, Rosenberg RD, Austen KF 1982 Structural determinants of the capacity of heparin to inhibit the formation of the human amplification convertase. J Clin Invest 67: 223–228.
Wegmuller E, Kazatchkine MD, Nydegger UE 1983 Complement activation during extracorporeal blood bypass. Plasma Ther Transfus Technol 4: 361–71.
Plotz FB, van Oeveren W, Hultquist KA, Miller C, Bartlett RH, Wildevuur Ch RH 1992 Heparin coated circuits reduce complement activation and release of inflammatory mediators during extracorporeal circulation in rabbits. Artif Organs 16: 366–370.
van Lingen RA, Hofhuis WDJ, Dekker I, Baerts W, Hahlen K, Sauer PJJ 1992 The effect of heparin in arterial catheters on the coagulation in preterm infants. J Perinat Med 20: 39–46.
Plotz FB, van Oeveren W, Bartlett RH, Wildevuur Ch RH 1993 Blood activation during neonatal extracorporeal life support (ECLS). J Thorac Cardiovasc Surg 105: 823–832.
Tracey KJ, Vlassara H, Cerami A 1989 Cachectin/tumor necrosis factor. Lancet 2: 1122–1125.
Rich EA, Panuska JR, Wallis RS, Wolf CB, Leonard ML, Ellner JL 1989 Dyscoordinate expression of tumor necrosis factor- by human blood monocytes and alveolar macrophages. Am Rev Respir Dis 130: 1010–1016.
Prescott SM, Zimmerman GA, McIntyre TM 1990 Platelet-activating factor. J Biol Chem 262: 4215–4222.
Caplan M, Hsueh W, Kelly A, Donovan M 1990 Serum PAF acetylhydrolase increases during neonatal maturation. Prostaglandins 39: 705–714.
Gaylord MS, Smith Z, Lorch V, Blank M, Anderson M, Snyder F 1990 Platelet-activating factor (PAF) levels in premature infants in the first two weeks of life as measured by a new radioimmunoassay (RIA). Pediatr Res 27: 205A
Sun X, Hsueh W 1991 Platelet-activating factor produces shock, in vivo complement activation, and tissue injury in mice. J Immunol 147: 509–514.
Schrod L, Frauendienst-Egger G, Stockhausen von HB, Kirschfink M 1992 Complement fragment C3a in plasma of asphyxiated neonates. Eur J Pediatr 151: 688–692.
Stimler NP, Hugli TE, Bloor CM 1980 Pulmonary injury induced by C3a and C5a anaphylatoxins. Am J Pathol 100: 327–348.
Zilow G, Zilow EP, Burger R, Linderkamp O 1993 Complement activation in newborn infants with early onset infection. Pediatr Res 34: 199–203.
Burger R, Zilow G, Bader A, Friedlein A, Naser W 1988 The C terminus of the anaphylatoxin C3a generated upon complement activation represents a neoantigenic determinant with diagnostic potential. J Immunol 141: 553–558.
de Bont ESJM, Martens A, van Raan J, Samson G, Fetter WPF, Okken A, de Leij LHFM 1993 Tumor necrosis factor-, interleukin-1, and interleukin-6 plasma levels in neonatal sepsis. Pediatr Res 33: 380–383.
Rubin BB, Smith A, Liauw S, Isenman D, Romaschin AD, Walker PM 1990 Complement activation and white cell sequestration in postischemic skeletal muscle. Am J Physiol 259:H525–H531.
MacPhaden AR, Whaley K 1985 The complement system in sepsis and trauma. Br Med Bull 41: 281–286.
Saugstad OD, Buo L, Johansen HT, Roise O, Aasen AO 1992 Activation of the plasma kallikrein-kinin system in respiratory distress syndrome. Pediatr Res 32: 431–435.
Brus F, van Oeveren W, Okken A, Bambang Oetomo S 1994 Activation of the plasma clotting, fibrinolytic and kinin-kallikrein system in preterm infants with severe idiopathic respiratory distress syndrome. Pediatr Res 36: 647–653.
Kaplan AP, Silverberg M 1987 The coagulation-kinin pathway of human plasma. Blood 70: 1–15.
Andrew M, Massicotte-Nolan P, Karpaatkin M 1983 Plasma protease inhibitors in premature infants: influence of gestational age, postnatal age, and health status. Proc Soc Exp Biol Med 173: 495–500.
Caplan MS, Kelly A, Hsueh W 1992 Endotoxin and hypoxia-induced intestinal necrosis in rats: the role of platelet-activating factor. Pediatr Res 31: 428–434.
Wirthmueller U, de Weck AL, Dahinden CA 1989 Platelet-activating factor production in human neutrophils by sequential stimulation with granulocyte macrophage colony-stimulating factor and the chemotactic factors C5a and formyl-methionylleucyl-phenylalanine. J Immunol 142: 3213–3218.
Acknowledgements
The authors thank Johan Haan for his technical assistance and Annalie van der Vijver, M.D., for correction of the manuscript.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Brus, F., Van Oeveren, W., Okken, A. et al. Activation of Circulating Polymorphonuclear Leukocytes in Preterm Infants with Severe Idiopathic Respiratory Distress Syndrome. Pediatr Res 39, 456–463 (1996). https://doi.org/10.1203/00006450-199603000-00013
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199603000-00013
This article is cited by
-
Heart rate and leukocytes after air and ground transportation in artificially ventilated neonates: a prospective observational study
Intensive Care Medicine (2009)
-
Deleted in Malignant Brain Tumors 1 (DMBT1) is present in hyaline membranes and modulates surface tension of surfactant
Respiratory Research (2007)
-
Activatie van plasma-eiwitten en bloedcellen bij het neonataal respiratoir distress-syndroom: pathogenetische en therapeutische aspecten
Tijdschrift voor kindergeneeskunde (2000)
