
Abstract
Forkheadbox protein 3 (FOXP3), initially identified as a key transcription factor for regulatory T cells (Treg cells), was also expressed in many tumors including pancreatic ductal adenocarcinoma (PDAC). However, its role in PDAC progression remains elusive. In this study, we utilized 120 PDAC tissues after radical resection to detect cancer-FOXP3 and Treg cells by immunohistochemistry and evaluated clinical and pathological features of these patients. Cancer-FOXP3 was positively correlated with Treg cells accumulation in tumor tissues derived from PDAC patients. In addition, high cancer-FOXP3 expression was associated with increased tumor volumes and poor prognosis in PDAC especially combined with high levels of Treg cells. Overexpression of cancer-FOXP3 promoted the tumor growth in immunocompetent syngeneic mice but not in immunocompromised or Treg cell-depleted mice. Furthermore, CCL5 was directly trans-activated by cancer-FOXP3 and promoted the recruitment of Treg cells from peripheral blood to the tumor site in vitro and in vivo. This finding has been further reinforced by the evidence that Treg cells recruitment by cancer-FOXP3 was impaired by neutralization of CCL5, thereby inhibiting the growth of PDAC. In conclusion, cancer-FOXP3 serves as a prognostic biomarker and a crucial determinant of immunosuppressive microenvironment via recruiting Treg cells by directly trans-activating CCL5. Therefore, cancer-FOXP3 could be used to select patients with better response to CCL5/CCR5 blockade immunotherapy.
Similar content being viewed by others


Introduction
Characterized by rapid progression, pancreatic ductal adenocarcinoma (PDAC) is one of the most malignant tumor with a 5-year survival rate <5%.1 The poor prognosis results from the early onset of invasion and metastasis, as well as lack of effective therapies. To improve PDAC survival rate, novel treatment modalities are urgently needed. In recent studies, immunotherapy has shown a promising effect on several solid tumors.2, 3, 4 PDAC tissues were initially defined as immune privileged milieu with rare effector T cells.5 Recently, several data from primary human tumor samples demonstrated that efficient cytotoxic (CD8+) T cells accumulated within the PDAC microenvironment and maintained the activity of secreting IFN-r.6, 7 The failure to control tumor growth may derive from multiple immunosuppressive mechanisms rather than deficiency of adaptive immunity.8
Infiltration of CD4+CD25+FOXP3+ regulatory T cells (Treg cells) is a major cause of immunologic tolerance and associated with poor prognosis in PDAC patients,9 but the mechanism for Treg cells infiltration in PDAC remains unclear.
Forkheadbox protein 3 (FOXP3) is a key transcription factor regulating development and function of Treg cells10 and its expression has been reported in several tumors such as melanoma, pancreatic cancer and breast cancer (cancer-FOXP3, referred as c-FOXP3 hereafter).11 However, the functions of c-FOXP3 are complicated and cell type dependent.12 C-FOXP3 has been initially defined as a tumor suppressor in breast, prostate and ovarian cancers through repressing the expression of several oncogenes and activating tumor-suppressor genes. On the contrary, c-FOXP3 has been identified as a biomarker of poor prognosis in urinary bladder cancer, non-small cell lung cancer, esophageal cancer and melanoma.13 In PDAC, a pilot study reported that c-FOXP3 was expressed in PDAC cells and suppressed the function of effector CD4+CD25-T cells by secreting transforming growth factor-β (TGF-β) in vitro.14 However, this pilot study is based on a small number of patients without follow-up (N=39) and limited to in vitro analysis. The clinical significance and function of c-FOXP3 in PDAC in vivo need to be elucidated.
In this study, we defined c-FOXP3 as a biomarker of poor prognosis based on the 120 samples of PDAC after radical resection. Intriguingly, c-FOXP3 was highly associated with the numbers of FOXP3+Treg cells, inspiring us to identify the possibility and mechanism of c-FOXP3 in recruiting Treg cells infiltration, which could help in optimizing the strategy of immunotherapy in PDAC.
Results
FOXP3 protein is overexpressed in human PDAC specimens and cell lines
A pilot study discovered the expression of FOXP3 in PDAC samples (defined as c-FOXP3), but its clinical significance was unclear. To better understand the role of c-FOXP3 in PDAC progression, immunohistochemistry was conducted to determine the FOXP3 expression in tumor tissues of 120 patients with PDAC. Normal pancreatic tissue, as well as the specimens of pancreatic tumors serous cystadenoma, pancreatic intraepithelial neoplasia and pancreatic neuroendocrine tumor was used as negative control. The specimen of the spleen was used as positive control. FOXP3 expression was detected in PDAC samples but not found in other samples (Figure 1a). In addition, normal pancreatic ductal epithelium cells adjacent to PDAC tissues were found negative for FOXP3 expression (Figure 1b). Intriguingly, robust expression of FOXP3 protein was found in both cytoplasm and nucleus of 76 PDAC tissues and was significantly correlated with shorter overall survival (OS; median time, 15 and 24 months, P<0.001) and relapse-free survival (RFS; median time, 9 and 15 months, P=0.0012; Figures 1c and d), suggesting that c-FOXP3 is a biomarker for poor prognosis of PDAC. FOXP3 expression was detected in four human PDAC cell lines, but showed negative in a normal pancreatic ductal epithelial cell line, which is consistent with the data from the tissue samples (Supplementary Figure 1A).

FOXP3 protein expression in PDAC cells and the clinical outcomes. (a) Immunohistochemical profiles (hematoxylin staining) of FOXP3 in normal pancreas, serous cystadenoma, pancreatic intraepithelial neoplasia (PanIN), pancreatic neuroendocrine tumor (PNET), spleen and PDAC tissues under microscopy. Magnification: × 200. (b) Expression of FOXP3 in normal pancreas cells (N) and PDAC (T) cells (brown). Magnification: × 40 and × 200. (c, d) Kaplan–Meier OS and RFS of PDAC patients, presented as high-c-FOXP3 or low-c-FOXP3 expression groups based on the log-rank statistic test (n=76 and n=44, respectively). Final scores of c-FOXP3 expression (1–9)=intensity score (1–3) × percentage score (1–3). High-c-FOXP3 >4 and low-c-FOXP3 ⩽4.
Prognostic impact of Treg cells accumulation and c-FOXP3 expression in PDAC
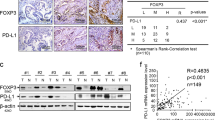
As FOXP3 is also associated with Treg cells, the number of FOXP3+Treg cells in tumor microenvironment was recorded and analyzed by immunohistochemistry. Intriguingly, FOXP3+Treg cells accumulated in the c-FOXP3 high-expression tissues, and the numbers of FOXP3+Treg cells were parallel to c-FOXP3 levels in PDAC cells (r=0.537, P<0.001; Figure 2a). It has been shown that FOXP3+immune cells in PDAC tissues mainly derived from CD4+T cells. To further detect the correlation between c-FOXP3 and CD4+CD25+FOXP3+Treg cells, immunofluorescence and flow cytometry analysis were performed on 10 fresh tissues of human PDAC. We found that PDAC tissues with higher c-FOXP3 expression showed higher accumulation of CD4+FOXP3+Treg cells, which matched the immunohistochemical results (Figures 2b and c). To understand the role of Treg cells in the clinical significance of c-FOXP3, patients with high c-FOXP3 were divided into two groups based on the Treg cells number (high Treg group ⩾10 Treg cells/high-power field and low Treg group <10 Treg cells/high-power field). As shown in Figure 2d, the medians of OS and RFS in the high c-FOXP3/high Treg cells group were significantly shorter than those in the high c-FOXP3/low Treg cells (14 and 6 months vs 18 and 10 months, respectively, P=0.01). Taken together, these results indicate that Treg cells accumulation has a critical role in the function of c-FOXP3 in PDAC patients.

c-FOXP3 correlates with Treg cells accumulation in PDAC microenvironment. (a) Counting FOXP3+Treg cells in PDAC microenvironment with low and high expression of c-FOXP3 protein (left). Statistical analysis of Treg cells accumulation in tumor with low and high expression of c-FOXP3 (right; n=120; **P<0.01) by Spearman's rank correlation test. High c-FOXP3 >4, low c-FOXP3 ⩽4. High Treg group ⩾10 Treg cells/high-power field (HPF), low Treg group <10 Treg cells/HPF). Magnification: × 200. (b) Immunofluorescence staining of FOXP3 expression and Treg cells accumulation in tumor tissues from fresh surgical samples (n=10). Magnification: × 200. (c) Flow cytometry analysis of Treg cells accumulation of the same samples. Summary data were shown below (mean±s.e.m., *P<0.05). (d) Kaplan–Meier OS and RFS for different levels of Treg cells accumulation with high c-FOXP3 expression based on the log-rank statistic test (P=0.01, P=0.03, respectively).
c-FOXP3 affecting tumor size in PDAC involves Treg cells accumulation
To identify the role of c-FOXP3 in the progression of PDAC, we analyzed the correlation between c-FOXP3 and several pathological parameters of PDAC patients. Interestingly, the patients with high c-FOXP3 showed larger tumor volumes than those with low c-FOXP3 (Figure 3a), whereas other parameters (including age, sex, lymphnode metastasis, pTNM stage and CA19-9) showed no significant differences (Supplementary Table 1). To identify the mechanism of c-FOXP3-induced tumor growth, we first evaluated whether c-FOXP3 directly affected the growth of PDAC. The stable cell lines with overexpressed or downexpressed FOXP3 were established (Supplementary Figure 1B). Apoptosis analysis, cell cycle analysis and Edu incorporation assay were performed, but none of them showed significant difference when FOXP3 was up or downregulated in these cell lines (Supplementary Figures 2A–C). Furthermore, SCID mice (immunocompromised) subcutaneously implanted with the Pan02-pLKO-FOXP3 cell line (the mouse pancreatic cancer cell line with FOXP3 knockdown) on the left abdomen and a Pan02-pLKO-Control cell line on the right abdomen also exhibited no difference on size, weight or proliferation between the two groups of tumors (Supplementary Figures 2D and E), suggesting that c-FOXP3 did not directly affect the growth of PDAC cells. In contrast, the volume and weight of the Pan02-pLKO-FOXP3 tumors implanted in the immunocompetent C57BL/6 mice were significantly smaller than those in the control group (P<0.05; Figure 3b, Supplementary Figure 3A). Reversely, the volume and weight of the Pan02-pLV-FOXP3 tumors were larger than those in the control group (P<0.05; Supplementary Figures 3B and C). FOXP3 and Ki67 staining of tumor sections suggested that knockdown or overexpression of c-FOXP3 may suppress or promote the proliferation of tumor cells in immunocompetent C57BL/6 mice, respectively (Figure 3c, Supplementary Figure 3D). Therefore, we propose that c-FOXP3-induced tumor growth is associated with the immunosuppressive effect. The hypothesis was supported by our data that Treg cells numbers in the c-FOXP3-interfered group varied from 4.5 to 9.6%, whereas that in the control group varied from 5.3 to 13.9% (P<0.05; Figure 3d). Similarly, Treg numbers in the c-FOXP3 overexpression group varied from 7.2 to 16.3%, whereas those in the control group varied from 5.8 to 9.3% (P<0.05; Supplementary Figure 3E). Meanwhile, along with the knockdown of c-FOXP3, CD8+T cells percentage in all CD45+leukocytes increased, whereas the percentage of total apoptotic CD8+T cells decreased (P<0.01; Figure 3e). After overexpression of c-FOXP3, CD8+T cells percentage in all CD45+leukocytes decreased, whereas the percentage of total apoptotic CD8+T cells increased (P<0.01; Supplementary Figure 3f). To further elucidate the role of Treg cells in tumor growth, six C57BL/6 mice received implantation of Pan02-pLKO-FOXP3 cells and Pan02-pLKO-Control cells in the right abdomen and the left abdomen, respectively, at day 0 (Figure 3e) and were treated with anti-mouse CD25 antibody injection for Treg cells depletion. Treg cells in peripheral blood were monitored at day 7 and day 28 and those in the tumor microenvironment were analyzed after the tumor harvest. As a result, CD4+CD25+FOXP3+Treg cells in both peripheral blood and tumors were almost completely depleted (Supplementary Figure 3G), and the tumors in the two groups showed no significant difference in both size and weight (Figure 3f). Collectively, the difference of the tumor size related to the expression of c-FOXP3 was found exclusively in the immunocompetent C57BL/6 mice, but not in the mice with Treg cell-depleted ones. This finding is consistent with our clinical data that c-FOXP3 expression alone did not affect tumor size in PDAC patients when Treg cells number was low in the tumor microenvironment. The group of high c-FOXP3/high Treg cells showed a larger tumor size than that of high c-FOXP3/low Treg cells (P=0.001; Figure 3g), suggesting that Treg cells accumulation mediates the tumor growth boosted by c-FOXP3 in PDAC.

c-FOXP3 affecting tumor size in PDAC involves Treg cells accumulation. (a) The real distribution of primary tumor size (T) between c-FOXP3 high and low expression groups (n=120, *P<0.05 by χ2 test. T: T1, tumor limited to the pancreas, 2 cm or less in greatest dimension. T2, tumor limited to the pancreas, >2 cm in greatest dimension. T3, Tumor extends beyond the pancreas but without involvement of the celiac axis or the superior mesenteric artery. (b) Experimental scheme for subcutaneous carcinoma model of C57BL/6 mice (top). Tumor volumes and weights for both groups of C57BL/6 mice (bottom). Pan02-pLKO-Control, n=8; Pan02-pLKO-FOXP3, n=8; *P<0.05 by paired Student’s t-test. (c) IHC staining of FOXP3 and Ki67 in both groups of tumor sections. Magnification: × 200. (d) Flow cytometry analysis of Treg cells accumulation in the tumor microenvironment of C57BL/6 murine model. Representative results were shown (left). Summary data were provided for each group except one undetectable because of operational factors (right, *P<0.05 by paired Student’s t-test). (e) Summary data of flow cytometry analysis of CD8+T cells percentage and apoptotic CD8+T cells in the tumor microenvironment of C57BL/6 murine model (**P<0.01 by paired Student’s t-test). (f) Experimental scheme for subcutaneous carcinoma model of C57BL/6 mice: CD25 antibody was injected at days -2, 1 and 21 intravenously. Tumor volume and weight of the C57BL/6 mice for both groups. (P>0.05 by paired Student’s t-test). (g) The distribution of the primary tumor size (T) among three c-FOXP3/Treg cells groups (only four cases exhibit low c-FOXP3/high Treg cells and fail to make statistical analysis. n=116, **P=0.001 by χ2 test).
c-FOXP3 recruits FOXP3+Treg cells in PDAC
The mechanisms of FOXP3+Treg cells accumulation may result from in situ proliferation, CD4+CD25-T cells conversion or recruitment from immune organs and peripheral blood. We first co-cultured FOXP3+Treg cells and pancreatic cancer cell lines with or without overexpression of c-FOXP3. IL-2 co-culture was used as positive control. Edu analysis showed that c-FOXP3 did not affect the proliferation of FOXP3+Treg cells (Supplementary Figure 4A). Second, we co-cultured CD4+CD25-Tcells and pancreatic cancer cell lines with or without overexpression of c-FOXP3. TGF-β was used as a positive control. Flow cytometry analysis suggested that c-FOXP3 did not affect the conversion of CD4+CD25-T cells toward Treg cells (Supplementary Figures 4B–E). Finally, an in vitro transwell model was setup to assess Treg cells recruitment toward PDAC cells. Treg cells migrated to the lower transwell chamber after 24 h were counted under microscope and analyzed by flow cytometry. As shown in Figures 4a and c, overexpression of c-FOXP3 strongly enhanced the recruitment of Treg cells in Panc-1, BxPC-3 and AsPC-1 cells. On the contrary, knockdown of c-FOXP3 significantly inhibited the recruitment of Treg cells in Panc-1, BxPC-3 and MIA PaCa-2 cells. In addition, Treg cells were isolated from peripheral blood mononuclear cells to reach the purity of 94.9% (Supplementary Figure 4F) and the total numbers of recruited Treg cells by pancreatic cancer cell lines were evaluated using the same method described above (Supplementary Figure 4G). In consistence with the results in the peripheral blood mononuclear cells recruitment assay, the expression level of c-FOXP3 was positively correlated to the numbers of Treg cells recruited to the lower chamber.

c-FOXP3 recruits Treg cells in vitro and in vivo. (a) FACS analysis of Treg cells recruitment by FOXP3 overexpressed Panc-1, BxPC-3, and AsPC-1 cells for 24- h relative to empty vector-transfected cells, and Treg cells recruitment by FOXP3-interfered Panc-1, BxPC-3 and MIA PaCa-2 cells for 24- h relative to control groups. Representative results were shown. (b) Percentage of recruited CD4+CD25+FOXP3+Treg cells from peripheral blood mononuclear cells (PBMCs; mean±s.e.m., n=5; *P<0.05 by Student’s t-test). (c) Total numbers of recruited Treg cells from PBMCs (mean±s.e.m., n=5; *P<0.05 by Student’s t-test). (d) Experimental scheme for orthotropic pancreatic carcinoma model: mice were divided into two groups (n=5 per group). One was implanted with Panc-1-pLV-Control cells and the other was implanted with Panc-1-pLV-FOXP3 cells. Both groups received human PBMCs injection on days 1, 8, 15, 22, 29 and 36 through tail vein. Tumors were harvested at the sixth week. (e) FACS analysis of Treg cells recruitment into tumor microenvironment of both groups was measured. Representative results were shown; (mean±s.e.m., n=5; *P<0.05 by Student’s t-test).
Furthermore, Treg cells recruitment was evaluated in vivo. We established an orthotropic implantation model in nude mice by injecting human pancreatic cancer cell lines of Panc-1-pLV-FOXP3 or Panc-1-pLV-Control into pancreas. The implanted nude mice were treated with human peripheral blood mononuclear cells injection through tail vein once a week. Tumors were harvested for analysis after 6 weeks (Figure 4d). Flow cytometry analysis showed that Treg cells infiltration in low-FOXP3 tumors (1.9 to 11.0%) is much lower than that in high-FOXP3 tumors (3.2 to 26.5%; P<0.05; Figure 4e). This is consistent with the results from in vitro experiments. In conclusion, c-FOXP3 has a drastic chemoattractant effect on Treg cells.
CCL5 expression correlates with c-FOXP3 levels in PDAC
After the recruitment of Treg cells by c-FOXP3 has been identified in vitro and in vivo, we try to find out the chemokines mediating the recruitment. Among the major recruitment-associated chemokines, CCL5 was found to change most significantly on the transcription level in response to FOXP3 overexpression or knockdown in Panc-1, Pan02, AsPC-1 and MIA PaCa-2 cell lines (Figure 5a, Supplementary Figure 5A). Western blot demonstrated that the CCL5 level was elevated in the FOXP3 overexpression cell lines and reduced in the FOXP3 knockdown cell lines (Figure 5b, Supplementary Figure 5B). Enzyme-linked immunosorbent assay further indicated that the secretion of CCL5 was elevated in the supernatant of FOXP3 overexpression cell lines and reduced in the FOXP3 knockdown cell lines (Figure 5c, Supplementary Figure 5C). In addition, c-FOXP3 was correlated and colocalized with CCL5 in the consecutive PDAC tumor tissues (P<0.001; Supplementary Table 2; Figure 5d). Thus, we propose that the expression and secretion of CCL5 is regulated by c-FOXP3 in pancreatic cancer cells.

c-FOXP3 directly activates CCL5 expression. (a) The mRNA level of CCL2, CCL3, CCL4, CCL5, CCL17, CCL20, CCL22 and CCL28 were measured by real-time–PCR (mean±s.e.m., n=3; *P<0.05 compared with Control, by Student’s t-test). (b) The protein level of c-FOXP3 and CCL5 in Panc-1 and Pan02 were detected by western blotting. (c) The secretion of CCL5 in the supernatant was detected by enzyme-linked immunosorbent assay (ELISA; mean±s.e.m., n=3; *P<0.05 compared with Control, by Student’s t-test). (d) c-FOXP3 and CCL5 expression in both normal pancreas and PDAC by immunohistochemistry. Magnification: × 200. (e) Specificity of the ChIP assay. Human CCL5 gene, including the FOXP3-binding motif 1–4 (upper). Binding of FOXP3 and CCL5 promoter was observed at motif-4. p21 was used as positive control (down). (f) The Panc-1 cells were transfected with either vector control or FOXP3 in conjunction with the luciferase reporter pGL3-CCL5-promoter (motif-4 WT) or pGL3-CCL5-promoter (motif-4MUT) vectors. pGL3-p21-promoter was used as positive control. After 48 hours, firefly and renilla luciferase activities were measured using the Dual-Luciferase Reporter assay (Promega, Madison, WI, USA) and the ratio was determined (*P<0.05, by Student’s t-test). The experiment was performed in triplicate and repeated three times with the same results.
c-FOXP3 directly activates CCL5 transcription in PDAC
To investigate whether CCL5 gene is a direct target of c-FOXP3, we screened out four potential FOXP3-binding sites in the human CCL5 promoter region (Figure 5e). Using P21 promoter as positive control, PCR was adopted to identify the binding motif isolated from chromatin immunoprecipitation (ChIP) assay performed in Panc-1 with the FOXP3 antibody. The ChIP complexes in Panc-1 cells showed the motif-4 have a strong affinity to FOXP3. To understand whether the binding of transcription factor FOXP3 with motif-4 could sufficiently trans-activate the CCL5 promoter, a pair of luciferase reporter vectors containing either the normal CCL5 promoter (–2100/–1) or the CCL5 promoter with mutated binding motif-4 were constructed. Luciferase assay showed that FOXP3 activated CCL5 expression by directly binding to the motif-4 in the CCL5 promoter (Figure 5f). Meanwhile, human and mice shared 85% homology in the amino-acid sequence of CCL5 (Supplementary Figure 5D) and seven forkhead-binding motifs were identified in the mouse CCL5 gene promoter. ChIP assay and luciferase assay performed in Pan02 cells showed that FOXP3 induced the transcription of mouse CCL5 (Supplementary Figures 5E and F). Taken together, we conclude that c-FOXP3 directly induces CCL5 transcription by binding to its promoter in pancreatic cancer cells in both human and mice.
CCL5 is involved in the recruitment of Treg cells by c-FOXP3 in PDAC
To further confirm that c-FOXP3 recruits Treg cells through CCL5, we performed a loss-of-function and gain-of-function assays by specific human CCL5 antibody blockade and human recombinant CCL5 treatment, respectively. As shown in Figures 6a and b, CCL5 antibody significantly reduced the Treg cells recruitment in FOXP3-overexpressed Panc-1 and AsPC-1 cells when compared with IgG control, whereas recombinant CCL5 promoted the Treg cells recruitment in FOXP3 knockdown MIA PaCa-2 and BxPC-3 cells when compared with saline control. Statistics showed significant differences in both the percentage and the cell count of the recruited Treg cells. To investigate the antagonistic effect of the CCL5 antibody to Treg cells recruitment in vivo, C57BL/6 mice were subcutaneously implanted with either Pan02-pLV-FOXP3 or Pan02-pLV-control cell lines on either side of the abdomen. Half of the mice from each implantation group were treated with either intratumoral or intraperitoneal injection of anti-mouse CCL5 antibody and the others were treated with IgG control (Figure 6c, Supplementary Figure 6A). After 28 days, all mice were killed for tumor harvest except for the one in the intratumoral group died after surgery. Flow cytometry analysis of the single-cell suspension indicated that Treg cells infiltration was decreased by blocking CCL5 in both Pan02-pLV-Control groups and Pan02-pLV-FOXP3 groups but has a stronger effect on the Pan02-pLV-FOXP3 groups (Figures 6d and e, Supplementary Figures 6B and C). Apoptotic CD8+T cells were also decreased along with the decreased Treg cells (Figure 6f, Supplementary Figure 6D). In addition, the average tumor volumes of the CCL5 antibody-treated groups were smaller than those of the IgG-treated groups. The growth inhibition rate in the Pan02-pLV-FOXP3 groups was more significant than that in the Pan02-pLV-Control groups (Figure 6g, Supplementary Figure 6E). In summary, CCL5 is involved in the recruitment of Treg cells by c-FOXP3, thereby promoting tumor growth in an immunosuppressive environment.

CCL5 is involved in the recruitment of Treg cells by c-FOXP3 in vitro and in vivo. (a) FACS analysis. Treg cells recruitment by Foxp3-overexpressed Panc-1 and AsPC-1 cell lines with the CCL5 neutralized antibody for 24-h relative to those with IgG. Treg cells recruitment by Foxp3 knockdown MIA PaCa-2 and BxPC-3 cell lines treated with recombinant CCL5 for 24-h relative to those with IgG. Percentages of recruited Treg cells from peripheral blood mononuclear cells (PBMCs) were shown (mean±s.e.m., n=3; *P<0.05 by Student’s t-test). (b) The cell count of the recruited Treg cells (mean±s.e.m., n=5; *P<0.05 by Student’s t-test). (c) Experimental scheme for subcutaneous carcinoma model of C57BL/6 mice: CCL5 antibody or isotype IgG was injected intratumoral (20 μg per mouse, twice a week). (d, e) FACS analysis of Treg cells recruitment into tumor microenvironment of the four groups was measured. Inhibition rate=(IgG-CCL5Ab) group/IgG group. (f) FACS analysis of CD8+T cells apoptosis in tumor microenvironment of the four groups was measured. (g) Tumor growth was evaluated by measuring tumor volumes and growth inhibition rate, and compared by one-way analysis of variance with Bonferroni post-hoc test (**P<0.01). Inhibition rate=(IgG-CCL5Ab) group/IgG group.
Discussion
It is well established that the immunosuppression caused by the accumulation of Treg cells in the tumor environment is a key factor for the poor prognosis in PDAC patients.15, 16, 17, 18 Recently, the discovery of FOXP3, a Treg cells marker, in a variety of cancers inspired the investigation to distinguish the function of c-FOXP3 from that of Treg-FOXP3. In this study, we identified that c-FOXP3 is critical to recruit Treg cells in the microenvironment of PDAC and CCL5, which is directly trans-activated by c-FOXP3, mediates the recruitment of FOXP3+T cells.
c-FOXP3 expression was initially identified in a cohort of 39 tissue samples from PDAC patients,14 but their study did not reveal the link between c-FOXP3 and pathological consequences because of the different clinical characteristics (including patients with curative and palliative operation) of recruited patients. In addition, the sample size was too small to analyze the role of c-FOXP3 in predicting prognosis. To better understand the clinical significance of c-FOXP3, we setup the reliable studies based on the tumor samples from 120 patients with radical resection and all the patients received the standard gemcitabine adjuvant therapy after operation.
In this cohort, we discovered that c-FOXP3 was overexpressed in PDAC tissues compared with normal pancreatic tissues and other types of pancreatic tumor. We also demonstrated that high c-FOXP3 predicted a poor prognosis in PDAC. In the tumor sections stained for FOXP3, we detected FOXP3+ tumor-infiltrating lymphocytes representing Treg cells, which is consistent with previous reports.14, 15 Intriguingly, the higher expression of c-FOXP3, the more Treg cells were found around the tumor microenvironment. As it has been identified that ~99% of FOXP3+T cells are CD4+Treg cells and <1% are CD8+Treg cells in tumor tissues,19 we sorted out the CD4+FOXP3+Treg cells from PDAC tissues and further evaluated the correlation between c-FOXP3 and CD4+Treg cells with flow cytometry and immunostaining. In addition, clinical data showed that in combination with the higher Treg cells level, the higher expression of c-FOXP3 was associated with worse prognosis, suggesting that c-FOXP3 exerted the pathological function, at least partially, through Treg cells accumulation. It should be noted that patients with higher c-FOXP3 expression exhibited shorter OS and RFS than those with lower c-FOXP3 expression even when the level of Treg infiltration was low. The shorter survival and quick relapse indicated that c-FOXP3 may have additional cancer-promoting functions in addition to recruiting Treg cells. Previous studies reported that c-FOXP3+ pancreatic cancer cells could directly inhibit the activity of effector T cells by secreting inhibitory cytokines.20 In cholangiocarcinoma, c-FOXP3 also functions to promote invasion and metastasis of cancer cells.21 Our ongoing study is recruiting PDAC patients in late stages and investigates the role of c-FOXP3 in metastasis of PDAC.
Previous studies suggested that c-FOXP3 directly boosted cell growth in other types of cancers.22 To investigate the mechanism of c-FOXP3 in promoting tumor growth, we first conducted a series of in vitro analysis to exclude the possibility that c-FOXP3 directly influenced cell growth. Then, we constructed cell lines with stable c-FOXP3 knockdown and planted them into mice models in different immune states. Data showed that knockdown of c-FOXP3 decreased the PDAC tumor size in the immunocompetent mice but not in the immunocompromised mice, suggesting that c-FOXP3-induced tumor growth is dependent on the immune system. Besides, c-FOXP3 did not promote tumor growth in Treg cell-depleted mice and it was only associated with tumor size when Treg cells number is high in PDAC samples. Taken together, Treg cells are involved in the tumor growth promoted by c-FOXP3 in PDAC.
Previous reports addressed the mechanism of how Treg cells affected tumor progression. In ovarian cancer, Treg cells infiltration led to poor prognosis by promoting angiogenesis in the tumor microenvironment.23 Moreover, Treg cells were found an anti-inflammatory role by inhibiting effector T cells, which may further promote immune escape.24 It was also reported that tumor-infiltrating Treg cells could induce apoptosis of effector T cells through killer molecules such as FasL, Granzyme B and TGF-β.25,26,27 In this study, we showed that knockdown of c-FOXP3 led to reduction of Treg cells and decreased the apoptosis of CD8+T cells, supporting that Treg cells could stimulate the proliferation of PDAC by inducing the apoptosis of tumor-infiltrating killer cells.
To understand whether Treg cells accumulation in PDAC is recruited by c-FOXP3-positive cancer cells, we performed transwell analysis and overexpression of FOXP3 in PDAC cell lines recruited more Treg cells in vitro. To exclude the possibility that increased Treg cells were derived from in situ proliferation, we cultured the Treg cells in the conditioned media from FOXP3 overexpressed PDAC cell lines, and they showed little proliferation under c-FOXP3 stimulation. Transformation of CD4+CD25- T cells into Treg cells under stimulation of tumor-derived TGF-β could also contribute to Treg cells accumulation.28, 29, 30 However, compared with normal PDAC cells, co-culture of naive CD4+CD25-T cells with FOXP3 overexpressed PDAC cells in the transwell system did not exhibit obvious induction (data not shown). Furthermore, in vivo analysis confirmed that c-FOXP3 could recruit Treg cells to the tumor microenvironment. Taken together, Treg cells accumulation in PDAC mainly results from the recruitment by c-FOXP3.
As the transwell system avoided the direct contact between PDAC cell lines and Treg cells, we proposed that the recruitment is mediated by the chemokines triggered by c-FOXP3. Several cytokines have been recognized as important media for Treg cells recruitment.31, 32 CCL28 was found to have a role in hypoxia-induced Treg cells recruitment in ovarian cancer.23 Meanwhile, it has been revealed that Treg cells were recruited through the pathway of TGF-β-miR-34a-CCL22 in hepatocellular carcinoma.33 CCL22 was discovered in Treg cells recruitment in breast cancer, which could be diminished by blocking CCR4.34 In pancreatic cancer, knockdown of CCR5 resulted in suppression of tumor growth and significant decrease of Treg cells in the tumor microenvironment, indicating that the CCL5/CCR5 pathway mediated the infiltration of Treg cells.35 In this study, we discovered for the first time that CCL5 was positively correlated with c-FOXP3 and directly trans-activated by c-FOXP3 in PDAC cells. When the CCL5/CCR5 signaling pathway was blocked by the CCL5 antibody in vitro and in vivo, Treg cells recruitment to the tumor microenvironment was reduced. The reduction rate is more significant in the c-FOXP3 high expression group, indicating that CCL5 is involved in the recruitment of Treg cells induced by c-FOXP3. Although CCR5 was also expressed in the effector T cells, its expression level was lower than in Treg cells.36 In combination with the finding that Treg cells exhibited a greater chemotactic response toward CCR5 than the effector T cells,37 it suggested a more important role of the CCL5/CCR5 pathway on Treg cells recruitment than the effector T cells in PDAC. Notably, the recruitment of Treg cells may not be solely driven by CCL5. Preliminary evidence suggested that other chemokine receptors, such as CCR4, CCR10 and CXCR4, were discovered on the cell surface of Treg cells.38, 39 Further investigation is needed to understand the synergistic effect of different chemokines in the recruitment of Treg cells in PDAC.
Treg cells depletion is a promising strategy to enhance antitumor immune response.40, 41, 42 Recently, a pilot clinical study showed a favorable response targeting FOXP3+CD4+Treg cells depletion in lung and esophageal carcinoma.43 However, unselective depletion of Treg cells may result in severe autoimmune disorders, impeding the clinical translation of this strategy. As CCL5 has been identified as a key chemokine for Treg cells infiltration in PDAC, it is more reasonable to develop tumor immunotherapy via blocking CCL5/CCR544, 45, 46 to reduce the Treg cells infiltration in the local site of PDAC. Combined with our findings, we propose that the patients with high c-FOXP3 expression may respond better to the strategy of CCL5/CCR5 blockade.
Recent evidence showed that multiple immunosuppressive compositions exist in the PDAC microenvironment including M2 (CD163) macrophages, the checkpoint signaling (PD-L1 and CTLA-4) and FOXP3+Treg cells, and combined absence of the immunosuppressive markers defined a subtype of PDAC with improved prognosis. As a single checkpoint inhibitor achieved only modest effect in the treatment of PDAC,47, 48 the combined blockade of immunosuppressive cells infiltration and the checkpoint signaling should be a promising strategy to better control PDAC.
It should be noted that there are several limitations for this study. First, to better understand the relationship between FOXP3+ tumor cells and FOXP3+Treg cells, we have to select sufficient tissues for calculating the numbers of Treg cells. Therefore, all the samples were from patients with radical resection. Thus, the role of c-FOXP3 in metastatic pancreatic cancer need to be further defined. Second, the interaction of c-FOXP3 and Treg cells should be complicated and organ specific. For instances, c-FOXP3 showed inverse correlation to the Treg cells numbers in colorectal cancer but no correlation to those in scirrhous gastric carcinoma,49 suggesting that the present findings were limited to pancreatic cancer.
In summary, we demonstrated that c-FOXP3 recruits FOXP3+Treg cells by directly activating the secretion of CCL5 in PDAC. The expression of c-FOXP3 together with infiltration of Treg cells correlates with the tumor size and poor prognosis. We discovered that selective depletion of Treg cells in the tumor microenvironment by CCL5 blockade may facilitate the antitumor immune response in PDAC patients, especially in those with high c-FOXP3 levels.
Materials and methods
Patients and tissue samples
This study included 120 patients undergoing classical R0 surgical resection with histological diagnosis of PDAC at Tianjin Medical University Cancer Institute and Hospital from January 2009 to October 2012. Patient samples were as follows: 76 males and 44 females; (median age=58 years, range 26–78 years). The pathological stages of patients were defined by the Union for International Cancer Control (UICC) based on the tumor-node-metastasis (TNM) classification, 24 patients demonstrated lymphatic metastasis and 92 patients were defined as TNM II. All participants enrolled in this study were of Chinese Han ethnicity. Ethical approval was obtained from the local ethics committee. Each participant gave us informed consent before participation. None of the patients had received chemotherapy or radiotherapy at the time when tissue samples were collected. All patients were treated with systemic gemcitabine-based chemotherapy after operation. Postoperative follow-up of patients were conducted every 3 months initially. OS was defined as the time interval from the date of surgery to that of death because of any cause or that of the last follow-up. RFS was calculated from the date of surgery to that of local recurrence or metastasis. Local recurrence or metastasis was diagnosed by radiological examination (contrast-enhanced computed tomography or magnetic resonance imaging scanning). By the time of the last follow-up (27 January 2015), 85 patients died of PDAC and 31 patients were still alive (4 patients were lost to follow-up). Normal pancreas tissues, pancreatic intraepithelial neoplasia tissues and pancreatic neuroendocrine tumor tissues were obtained from adult patients who received operation for the diseases other than PDAC. Cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and tested by short tandem repeat profiling before application. Details were shown in the Supplementary materials and methods.
Mice
Female SCID mice, nude mice and C57BL/6 mice of 4 weeks were used in animal experiments. All mice were maintained in the specific pathogen-free conditions and animal experiment procedures were approved by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital, in compliance with the principles and procedures of the NIH Guide for the Care and Use of Laboratory Animals. All groups were randomly divided. The CD25 antibody or the isotype control (0.5 mg per mouse, PC61, BioXcell, West Lebanon, NH, USA) was injected on days –2, 1 and 21 following the previous literature with modifications.18 The CCL5 antibody or the isotype control (20 μg intratumorally per mouse, R&D Systems, Minneapolis, MN, USA) was injected twice a week. Harvested tumors were processed into the single-cell suspension with 1 mg/ml collagenase, 2.5 U/ml hyaluronidase and 0.1 mg/ml DNase. Details were shown in the Supplementary materials and methods.
Immunohistochemistry and immunofluorescence
PDAC specimens were processed with formalin fixation, paraffin embedding and sectioned to 5 μm in thickness. Immunohistochemistry was performed with a standard protocol. Immunofluorescence was performed with frozen samples. Antibodies used are listed in Supplementary Table 3. Details were shown in the Supplementary materials and methods.
Quantification of FOXP3+T cells
Immunohistochemistry for FOXP3+T cells was performed using standard procedures. Treg cells were counted under microscopy. Ten high-power fields (at × 40 magnification) were randomly selected from each specimen for digital photographs in area of 0.0625 mm2. Total numbers of lymphoid infiltrates and Treg cells in each field were counted manually by two investigators who did not know the clinical pathological characteristics and outcomes of patients.
Treg cells recruitment in vitro
Treg cells/peripheral blood mononuclear cells were placed in the upper chamber of the transwell and pancreatic cancer cells were placed in the lower chamber. After 24-h co-culture, cells in the lower chamber were collected and analyzed by flow cytometry for Treg cells population.
Western blot
Total protein was extracted with sodium dodecyl sulfate lyses buffer containing the proteinase inhibitors cocktail (Sigma, St Louis, MO, USA). Electrophoresis was performed as described in a standard protocol. Antibodies used are listed in Supplementary Table 3. Details were shown in the Supplementary materials and methods.
Chromatin immunoprecipitation
ChIP assay was performed according to the manufacturer’s instruction of a commercial kit (Millipore Corp., Bedford, MA, USA). PCR primers were listed in Supplementary Table 4.
Quantitative real-time PCR
Total RNA was extracted with TRIZOL Reagent (Invitrogen, Carlsbad, CA, USA). Complementary DNA was prepared by reverse-transcription as standard protocol described, followed by quantitative real-time PCR. Primers were listed in Supplementary Table 4.
Enzyme-linked immunosorbent assay
PDAC cell lines (1 × 107cells) were cultured for 72 h and the conditioned media collected after centrifugation at 700 g for 5 min at 4 °C. CCL5 protein was quantified using human CCL5 ELISA kit (R&D Systems) according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed with SPSS 18.0 software (SPSS Inc., Chicago, IL, USA) and GraphPad prism version 5.0 for Windows (GraphPad Software, San Diego, CA, USA). Paired/unpaired Student’s t-test, one-way analysis of variance test, Spearman rank correlation coefficient test, χ2 tests, Kaplan–Meier survival curves and log-rank test were performed. P<0.05 was considered significant.
References
Siegel RL, Miller KD, Jemal A . Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5–29.
Foley K, Kim V, Jaffee E, Zheng L . Current progress in immunotherapy for pancreatic cancer. Cancer Lett 2016; 381: 244–251.
Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res 2010; 16: 5067–5078.
Generali D, Bates G, Berruti A, Brizzi MP, Campo L, Bonardi S et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin Cancer Res 2009; 15: 1046–1051.
Vonderheide RH, Bayne LJ . Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol 2013; 25: 200–205.
Pillarisetty VG . The pancreatic cancer microenvironment: an immunologic battleground. Oncoimmunology 2014; 3: e950171.
Zhao T, Ren H, Jia L, Chen J, Xin W, Yan F et al. Inhibition of HIF-1alpha by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 2015; 6: 2250–2262.
Lutz ER, Kinkead H, Jaffee EM, Zheng L . Priming the pancreatic cancer tumor microenvironment for checkpoint-inhibitor immunotherapy. Oncoimmunology 2014; 3: e962401.
Yamamoto T, Yanagimoto H, Satoi S, Toyokawa H, Hirooka S, Yamaki S et al. Circulating CD4(+)CD25(+) regulatory T cells in patients with pancreatic cancer. Pancreas 2012; 41: 409–415.
Josefowicz SZ, Lu LF, Rudensky AY . Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30: 531–564.
Karanikas V, Speletas M, Zamanakou M, Kalala F, Loules G, Kerenidi T et al. Foxp3 expression in human cancer cells. J Transl Med 2008; 6: 19.
Martin F, Ladoire S, Mignot G, Apetoh L, Ghiringhelli F . Human FOXP3 and cancer. Oncogene 2010; 29: 4121–4129.
Triulzi T, Tagliabue E, Balsari A, Casalini P . FOXP3 expression in tumor cells and implications for cancer progression. J Cell Physiol 2013; 228: 30–35.
Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res 2007; 67: 8344–8350.
Hiraoka N, Onozato K, Kosuge T, Hirohashi S . Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 2006; 12: 5423–5434.
Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Brit J Cancer 2013; 108: 914–923.
Tang YC, Xu XJ, Guo SX, Zhang CB, Tang Y, Tian Y et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. Plos One 2014; 9: e91551.
Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC et al. Activated pancreatic stellate cells sequester CD8(+) T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013; 145: 1121–1132.
Kryczek I, Liu R, Wang G, Wu K, Shu X, Szeliga W et al. FOXP3 defines regulatory T cells in human tumor and autoimmune disease. Cancer Res 2009; 69: 3995–4000.
Hinz S, Pagerols-Raluy L, Oberg HH, Ammerpohl O, Grussel S, Sipos B et al. Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Res 2007; 67: 8344–8350.
Ma C, Peng C, Lu X, Ding X, Zhang S, Zou X et al. Downregulation of FOXP3 inhibits invasion and immune escape in cholangiocarcinoma. Biochem Biophys Res Commun 2015; 458: 234–239.
Heinze E, Baldwin S, Chan G, Hansen J, Song J, Clements D et al. Antibody-mediated FOXP3 protein therapy induces apoptosis in cancer cells in vitro and inhibits metastasis in vivo. Int J Oncol 2009; 35: 167–173.
Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011; 475: 226–230.
Facciabene A, Motz GT, Coukos G . T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res 2012; 72: 2162–2171.
Vignali DA, Collison LW, Workman CJ . How regulatory T cells work. Nat Rev Immunol 2008; 8: 523–532.
Lin YC, Chang LY, Huang CT, Peng HM, Dutta A, Chen TC et al. Effector/memory but not naive regulatory T cells are responsible for the loss of concomitant tumor immunity. J Immunol 2009; 182: 6095–6104.
Strauss L, Bergmann C, Whiteside TL . Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol 2009; 182: 1469–1480.
Zou W . Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006; 6: 295–307.
Moo-Young TA, Larson JW, Belt BA, Tan MC, Hawkins WG, Eberlein TJ et al. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J Immunother 2009; 32: 12–21.
Li X, Ye F, Chen H, Lu W, Wan X, Xie X . Human ovarian carcinoma cells generate CD4(+)CD25(+) regulatory T cells from peripheral CD4(+)CD25(-) T cells through secreting TGF-beta. Cancer Lett 2007; 253: 144–153.
Qin XJ, Shi HZ, Deng JM, Liang QL, Jiang J, Ye ZJ . CCL22 recruits CD4-positive CD25-positive regulatory T cells into malignant pleural effusion. Clin Cancer Res 2009; 15: 2231–2237.
Trujillo G, Hartigan AJ, Hogaboam CM . T regulatory cells and attenuated bleomycin-induced fibrosis in lungs of CCR7-/- mice. Fibrogenesis Tissue Repair 2010; 3: 18.
Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012; 22: 291–303.
Menetrier-Caux C, Faget J, Biota C, Gobert M, Blay JY, Caux C . Innate immune recognition of breast tumor cells mediates CCL22 secretion favoring Treg recruitment within tumor environment. Oncoimmunology 2012; 1: 759–761.
Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol 2009; 182: 1746–1755.
Cavassani KA, Campanelli AP, Moreira AP, Vancim JO, Vitali LH, Mamede RC et al. Systemic and local characterization of regulatory T cells in a chronic fungal infection in humans. J Immunol 2006; 177: 5811–5818.
Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, Piccirillo CA . CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med 2006; 203: 2451–2460.
Bayry J, Tchilian EZ, Davies MN, Forbes EK, Draper SJ, Kaveri SV et al. In silico identified CCR4 antagonists target regulatory T cells and exert adjuvant activity in vaccination. Proc Natl Acad Sci USA 2008; 105: 10221–10226.
Righi E, Kashiwagi S, Yuan JP, Santosuosso M, Leblanc P, Ingraham R et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res 2011; 71: 5522–5534.
Pastille E, Bardini K, Fleissner D, Adamczyk A, Frede A, Wadwa M et al. Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer. Cancer Res 2014; 74: 4258–4269.
Wang Y, Sparwasser TL, Figlin R, Kim HL . Combination of Foxp3+T cells depletion and mTOR inhibitor enhances immune memory and controls cancer growth. Cancer Res 2014; 74: 2217–2228.
deLeeuw RJ, Kost SE, Kakal JA, Nelson BH . The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res 2012; 18: 3022–3029.
Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T et al. Phase Ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res 2015; 21: 4327–4336.
Ward ST, Li KK, Hepburn E, Weston CJ, Curbishley SM, Reynolds GM et al. The effects of CCR5 inhibition on regulatory T-cell recruitment to colorectal cancer. Br J Cancer 2015; 112: 319–328.
Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol 2012; 189: 5602–5611.
Borczuk AC, Papanikolaou N, Toonkel RL, Sole M, Gorenstein LA, Ginsburg ME et al. Lung adenocarcinoma invasion in TGF beta RII-deficient cells is mediated by CCL5/RANTES. Oncogene 2008; 27: 557–564.
Medema RH, Macurek L . Checkpoint control and cancer. Oncogene 2012; 31: 2601–2613.
Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010; 33: 828–833.
Yoshii M, Tanaka H, Ohira M, Muguruma K, Iwauchi T, Lee T et al. Expression of Forkhead box P3 in tumour cells causes immunoregulatory function of signet ring cell carcinoma of the stomach. Br J Cancer 2012; 106: 1668–1674.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants 81272685, 81525021, 81672431, 81672435, 81502067, 81302082, 31471340, 31470957, 81401957 and 81472264), the National Key Clinical Specialist Construction Programs of China (no. 2013-544) and Key Program of Public Health Bureau Foundation of Tianjin (15KG 144).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Wang, X., Lang, M., Zhao, T. et al. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3+Treg cells in pancreatic ductal adenocarcinoma. Oncogene 36, 3048–3058 (2017). https://doi.org/10.1038/onc.2016.458
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.458
This article is cited by
-
IRF4-mediated Treg phenotype switching can aggravate hyperoxia-induced alveolar epithelial cell injury
BMC Pulmonary Medicine (2024)
-
Tumor immune microenvironment-based therapies in pancreatic ductal adenocarcinoma: time to update the concept
Journal of Experimental & Clinical Cancer Research (2024)
-
Acute myeloid leukemia-derived exosomes deliver miR-24-3p to hinder the T-cell immune response through DENN/MADD targeting in the NF-κB signaling pathways
Cell Communication and Signaling (2023)
-
Molecular and metabolic regulation of immunosuppression in metastatic pancreatic ductal adenocarcinoma
Molecular Cancer (2023)
-
Transcriptional control of pancreatic cancer immunosuppression by metabolic enzyme CD73 in a tumor-autonomous and -autocrine manner
Nature Communications (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}