
Abstract
We developed Copy Number Segmentation by Regression Tree in Next Generation Sequencing (CONSERTING), an algorithm for detecting somatic copy-number alteration (CNA) using whole-genome sequencing (WGS) data. CONSERTING performs iterative analysis of segmentation on the basis of changes in read depth and the detection of localized structural variations, with high accuracy and sensitivity. Analysis of 43 cancer genomes from both pediatric and adult patients revealed novel oncogenic CNAs, complex rearrangements and subclonal CNAs missed by alternative approaches.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Mullighan, C.G. et al. N. Engl. J. Med. 360, 470–480 (2009).
Ley, T.J. et al. Nature 456, 66–72 (2008).
Chiang, D.Y. et al. Nat. Methods 6, 99–103 (2009).
Xie, C. & Tammi, M.T. BMC Bioinformatics 10, 80 (2009).
Boeva, V. et al. Bioinformatics 27, 268–269 (2011).
Abyzov, A., Urban, A.E., Snyder, M. & Gerstein, M. Genome Res. 21, 974–984 (2011).
Xi, R. et al. Proc. Natl. Acad. Sci. USA 108, E1128–E1136 (2011).
Downing, J.R. et al. Nat. Genet. 44, 619–622 (2012).
Zhang, J. et al. Nature 481, 157–163 (2012).
Roberts, K.G. et al. Cancer Cell 22, 153–166 (2012).
Zhang, J. et al. Nature 481, 329–334 (2012).
Zhang, J. et al. Nat. Genet. 45, 602–612 (2013).
Brennan, C.W. et al. Cell 155, 462–477 (2013).
Pleasance, E.D. et al. Nature 463, 191–196 (2010).
Wang, J. et al. Nat. Methods 8, 652–654 (2011).
Stephens, P.J. et al. Cell 144, 27–40 (2011).
Sanborn, J.Z. et al. Cancer Res. 73, 6036–6045 (2013).
Handsaker, R.E., Korn, J.M., Nemesh, J. & McCarroll, S.A. Nat. Genet. 43, 269–276 (2011).
Parker, M. et al. Nature 506, 451–455 (2014).
Wu, G. et al. Nat. Genet. 46, 444–450 (2014).
Li, H. et al. Bioinformatics 25, 2078–2079 (2009).
Edmonson, M.N. et al. Bioinformatics 27, 865–866 (2011).
Breiman, L., Friedman, J.M., Olshen, R. & Stone, C. Classification and Regression Trees edn. 1 (Chapman and Hall/CRC, 1984).
Schwarz, G. Ann. Stat. 6, 461–464 (1978).
Kent, W.J. et al. Genome Res. 12, 996–1006 (2002).
Rozen, S. & Skaletsky, H. Methods Mol. Biol. 132, 365–386 (2000).
Acknowledgements
This study was supported by the St. Jude Children's Research Hospital–Washington University Pediatric Cancer Genome Project, Cancer Center support grant P30 CA021765 from the US National Cancer Institute and the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital. C.G.M. is supported as a Pew Scholar in the Biomedical Sciences and is a St. Baldrick's Scholar.
Author information
Authors and Affiliations
Contributions
X.C. and J.Z. conceived and designed the CONSERTING algorithm. X.C., P.G. and J.W. implemented the algorithm. J.Z., S.J.B., M.A.D., J.R.D., D.W.E. and C.G.M. designed the experiment. X.C., J.W., J.D.D., M.P., J.M., M.R., G.W., A.P., S.E., S. Pounds and J.Z. analyzed the data. K.R., J.D.D., S. Patel, L.H., D.P. and J.E performed validation and functional assays. J.N. and S.S. generated COLO-829 whole-genome sequencing data. X.C. and J.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
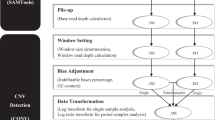
Supplementary Figure 1 Overview of the CONSERTING process.
A parallelogram shows input or output files, and a rectangle defines an analytical process. A diamond defines the condition for a follow-up process.
Supplementary Figure 2 CIRCOS plots for CNAs derived from SNP array, CONSERTING, CNV-seq, SegSeq, FREEC and BIC-seq for the 20 paired tumor-normal whole-genome sequencing data sets presented in this paper (SegSeq was not run on SJTALL015 and SJLGG039, as these two samples were mapped to hg19, which is not compatible with SegSeq).
All CIRCOS plots are shown in the same order. (a) Legend for the CIRCOS plot. (b) CIRCOS plots for the 12 ETP-ALL cases. (c) CIRCOS plots for the four RB cases. (d) CIRCOS plots for two B-ALL samples from COG. (e) A CIRCOS plot for the non-ETP T-ALL sample SJTALL015. (f) A CIRCOS plot for the low-grade glioma sample SJLGG039 (arrows show amplifications identified by both CONSERTING and SNP array).
Supplementary Figure 3 Somatic CNAs computed by CONSERTING and BIC-seq for 22 paired whole-genome sequencing data sets from The Cancer Genome Atlas (TCGA) GBM project.
Each sample is displayed in a colored box with the BIC-seq results (denoted by B) shown at the top, the SNP array result (denoted by S, and downloaded from TCGA) at the middle and the CONSERTING results (denoted by C) at the bottom. TCGA sample I.D.s are at the left. Samples marked with an asterisk had lower than median F1 scores for both CONSERTING and BIC-seq. Diagnosis and relapse sample pairs are shown in same color with different intensities. Pairs with underlined sample I.D.s had highly divergent CNA profiles from diagnosis to relapse.
Supplementary Figure 4 ROC curves for CONSERTING and BIC-seq in 11 ETP-ALL samples.
SJTALL008 was excluded from this analysis because there were no CNV calls in the curated SNP array result.
Supplementary Figure 5 CNA calls at chr1:164 -244 Mb of retinoblastoma tumor SJRB003 by SNP array, CONSERTING and four other methods.
(a) Global view of the CNA state in the 93-Mb region on 1q. The thin horizontal lines define the copy-neutral state (i.e., no copy-number variation). Blue blocks above the “neutral” line are the copy-number gains (amplifications) identified by each method, with the height of the block corresponding to the amplitude of the copy-number gain. (b) Detailed view of a 35-kb region showing that BIC-seq missed two breakpoints separating two CNV segments. Both breakpoints were confirmed experimentally by Sanger sequencing. The two CNV boundaries at chr1:231118034 and chr1:231123028 missed by BIC-seq are part of the complex rearrangement depicted in Supplementary Fig. 5c. Both were involved in interchromosomal translocations 60–70 Mb upstream, and the breakpoints were experimentally validated. (c) A complex rearrangement identified by CONSERTING at chr1:164-244 Mb. The top panel is an SV graph that connects the 12 SVs identified in this region. The black lines mark the breakpoints of the seven SVs detected only by CONSERTING, and the gray lines mark the breakpoints of the five SVs detected by both CONSERTING and CREST. The purple bar marks the boundaries of the CNA segment, with the amplitudes of CNAs marked at the bottom. The three colored dots mark the location of FISH probes used to validate the copy number and SVs. (d) PCR amplicon validation of CONSERTING-predicted SVs. Lane 68 (predicted amplicon size: 500 bp): chr1:231123028(-)|chr1:164744054(+); lane 69 (predicted amplicon size: 313 bp): chr1:166476222(+)|chr1:174088270(+); Lane 70 (predicted amplicon size: 343 bp): chr1:173901001(+)|chr1:233328796(+); Lane 71 (predicted amplicon size: 488 bp): chr1:230868636(-)|chr1:173478509(+); lane 72 (predicted amplicon size: 332 bp): chr1:241160552(+)|chr1:177362495(-); lane 73 (predicted amplicon size: 350 bp): chr1:177464060(+)|chr1:224896416(+); lane 74 (predicted amplicon size: 371 bp): chr1:236474589(+)|chr9:136321446(-). (e) FISH validation of the WGS predicted gain (scale bar, 10 µm; also refer to Supplementary Fig. 14 of Zhang, J. et al. Nature 481, 329–334 (2012)).
Supplementary Figure 6 Double-minute chromosomes identified in the TCGA-GBM data set.
The inner green and magenta lines connect intra- and interchromosomal SV breakpoints, respectively. The red and blue arcs represent amplification and deletion identified by CONSERTING, respectively. The thickness of the arc is proportional to the level of amplification or deletion. Samples marked with an asterisk (06-0152-01A, 06-0210-01A, 06-0211-01A, 06-0211-02A, 06-0648-01A,14-1402-01A, 14-1402-02A, 19-5960-01A and 27-1831-01A) had a chromothripsis-like CNA-SV profile.
Supplementary Figure 7 A novel intragenic NOTCH1 deletion resulting in expression of ICN T-ALL.
(a) The normalized read depth for part of NOTCH1 for normal germline (blue), tumor (red) and tumor-normal (gray), showing a deletion spanning exons 14–27 in the tumor sample. (b) A deletion in this region detected by CONSERTING but not by SNP array with two ends mapped within exons 14 and 27. (c) Sanger-sequencing chromatograms confirming the in-frame deletion in both genomic DNA and cDNA. (d) Western blot analysis of SJTALL015 with the intragenic deletion (∆) targeting the NOTCH1 HD domain, and the T cell lines MOLT3, HPBALL, DND41, PF382, TALL-1 and LOUCY and the murine fibroblast cell line GPE-86, either wild type (-) or harboring activating mutations (+). (e) NOTCH1 domain view showing that the intragenetic deletion removes several EGF domains, an LNR domain and the heterodimerization (HD) domain.
Supplementary Figure 8 Fractured genome in WGS data from SJRB002 and data from The Cancer Genome Atlas (TCGA) project.
(a) CNA plots of chromosomes 5 and 6 using the initial WGS data. Copy-number gain with log2 ratio > 0.17 is marked in red, copy-number loss with log2 ratio < –0.17 is marked in blue, and the remaining segments are marked in black. (b) CNA plot of the same sample based on the second WGS data set with no ‘fracture’. The 6p amplification is the only CNA that is replicated in the two WGS data sets (not detected by SNP array but validated by SKY mapping). (c) Genome-wide CNAs using the original WGS by all methods. (d) Genome-wide CNAs using the second 10x WGS by all methods. (e) Number of predicted somatic CNAs for TCGA-GBM samples with and without fractured genome by BIC-seq, CONSERTING and SNP array. (f) Three paired WGS data showing fractured genome. Each sample is displayed in a colored box with the BIC-seq results shown at the top, the SNP array result (downloaded from TCGA) at the middle and the CONSERTING results at the bottom. TCGA sample I.D.s are at the left. Gain is shown in red, and loss in blue.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–9 and Supplementary Data 1–4 (PDF 1523 kb)
Supplementary Table 1
Sample characteristics of 43 tumor pairs. (XLS 36 kb)
Supplementary Table 2
Manually curated autosomal CNAs by SNP array analysis in 12 ETP-ALL tumors. (XLS 41 kb)
Supplementary Table 3
Comparison of CNAs from whole-genome sequencing by CONSERTING, BIC-seq, SegSeq, CNV-seq and FREEC with manually curated CNVs from SNP array analysis in the 12 ETP-ALL samples. (XLS 49 kb)
Supplementary Table 4
Comparison of CNAs from whole-genome sequencing by CONSERTING and BIC-seq with CNAs from SNP array analysis in the 22 TCGA-GBM samples. (XLS 53 kb)
Supplementary Table 5
CNA profile and SV matching status of diluted COLO-829 sample analyzed by CONSERTING. (XLS 49 kb)
Supplementary Table 6
Experimental validation of novel CNAs in coding exons identified by CONSERTING and SegSeq in the two COG samples and SJTALL015. (XLS 36 kb)
Supplementary Software
CONSERTING package including instructions (README.pdf) and source code. Please see http://www.stjuderesearch.org/site/lab/zhang for updates, test data and instructions for running CONSERTING on the Amazon Web Services cloud (ZIP 1593 kb)
Rights and permissions
About this article

Cite this article
Chen, X., Gupta, P., Wang, J. et al. CONSERTING: integrating copy-number analysis with structural-variation detection. Nat Methods 12, 527–530 (2015). https://doi.org/10.1038/nmeth.3394
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.3394
This article is cited by
-
Atypical teratoid/rhabdoid tumoroids reveal subgroup-specific drug vulnerabilities
Oncogene (2023)
-
Genetic and epigenetic features of bilateral Wilms tumor predisposition in patients from the Children’s Oncology Group AREN18B5-Q
Nature Communications (2023)
-
RNAseqCNV: analysis of large-scale copy number variations from RNA-seq data
Leukemia (2022)
-
The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms
Nature Communications (2021)
-
Comprehensive molecular characterization of pediatric radiation-induced high-grade glioma
Nature Communications (2021)
