
Abstract
Most of the peptides presented by major histocompatibility complex (MHC) class I molecules require processing by proteasomes. Tripeptidyl peptidase II (TPPII), an aminopeptidase with endoproteolytic activity, may also have a role in antigen processing. Here, we analyzed the processing and presentation of the immunodominant human immunodeficiency virus epitope HIV-Nef(73–82) in human dendritic cells. We found that inhibition of proteasome activity did not impair Nef(73–82) epitope presentation. In contrast, specific inhibition of TPPII led to a reduction of Nef(73–82) epitope presentation. We propose that TPPII can act in combination with or independent of the proteasome system and can generate epitopes that evade generation by the proteasome-system.
Similar content being viewed by others


Main
The ability of an organism to eliminate viral infections relies largely on its capacity to generate peptides from viral antigens, which, in the context of major histocompatibility complex (MHC) class I molecules, can be presented on the cell surface to CD8+ cytotoxic T lymphocytes (CTLs). To allow binding to MHC molecules, intracellular proteins must be processed to smaller fragments, which are translocated by TAP (transporter associated with antigen processing) into the endoplasmic reticulum (ER). Finally, peptides of 8–11 residues in length, called epitopes, containing an appropriate binding motif bind to MHC class I molecules for transportation to the cell surface1,2,3,4,5. Most peptides that bind to MHC class I molecules are generated from cellular proteins by the 26S ubiquitin-proteasome system, the major proteolytic machinery in the cytosol5. Its proteolytic activity is exerted by the 20S core proteasome1,3 by three of the seven β-subunits in the two inner rings of the four-ring particle. The 20S core proteasomes are usually found associated with a 19S regulatory complex that binds to the outer α-rings of the 20S core to form the 26S proteasome. This complex is responsible for the binding of ubiquitin-tagged substrates and transportation of the substrates into the 20S core complex for processing. Stimulation of cells with interferon (IFN)-γ results in an exchange of the catalytic subunits, the formation of so-called immunoproteasomes and an adaptation of the proteasome to the specific requirements of an enhanced cellular immune response1,3,5.
Apart from its ability to generate peptides of the appropriate length, the central role of the proteasome in the antiviral CD8+ T cell–dependent immune response is largely based on its intrinsic ability to efficiently generate the C-terminal anchor residue of an epitope, which allows its proper binding to the peptide binding groove of the MHC class I protein1,3,5. Many epitopes are generated as precursor peptides that carry the correct C terminus and an N-terminal extension of several residues3,6,7,8. These epitope precursor peptides require N-terminal trimming by aminopeptidases either in the ER9,10,11,12 or in the cytosol13,14.
MHC class I–associated epitope generation from the Nef protein encoded by human immunodeficiency virus 1 (HIV-1 Nef) has been extensively studied, as this protein may be a good target for vaccination against AIDS. The Nef protein is expressed early and its processed MHC class I epitopes are recognized by CD8+ T lymphocytes on the cell surface before structural proteins are synthesized. Thus, infected cells could potentially be lysed and viral replication inhibited before virus release. The proteasome is known to produce a substantial number of MHC class I–restricted epitopes from HIV-1 Nef in the context of different HLA haplotypes15,16. However, production of the HIV Nef epitope from amino acids 73-81 (HIV Nef (73-82)) that is restricted to both HLA-A3 and HLA-A11 (HLA-A3/A11) appeared to be insensitive to proteasome inhibition16. In agreement with this, it had been suggested that epitopes carrying a lysine residue at its C terminus, as is the case for HIV Nef(73–82), may be generated by proteasomes with strongly reduced efficiency17. These data indicated that there may be additional proteolytic pathways that are involved in the cytosolic processing of MHC class I epitopes.
A protease that was previously suggested to also have a role in MHC class I antigen processing is the cytosolic subtilisin-like tripeptidyl peptidase II (TPPII)18,19. Due to its aminopeptidase activity, TPPII was suggested to function as a post-proteasomal trimpeptidase for epitope precursor molecules20. In addition to its exo-peptidase activity, TPPII also exhibits endo-proteolytic cleavage properties and it is able to cleave after lysine residues19. Thus, TPPII may be a candidate protease for the generation of those epitopes that cannot be produced by the proteasome.
We undertook these studies to identify the proteases that were essential for the generation of the HLA-A3/A11-restricted HIV Nef(73–82) epitope (QVPLRPMTYK) in human dendritic cells (DCs) expressing the full-length Nef protein. DCs are probable ports of entry for HIV during mucosal infection, which is the most frequent transmission mode, and they are the only antigen-presenting cells (APCs) that can stimulate naive T lymphocytes (that is, at the onset of infection as well as for vaccination). Using biochemical and immunological approaches, we found that purified 20S or 26S proteasomes were unable to generate the HIV Nef(73–82) epitope in vitro from larger synthetic polypeptides or from an ODC-Nef fusion protein, and that specific inhibition of proteasomes in human DCs did not affect epitope presentation. We found that purified high-molecular weight TPPII generates the HIV Nef(73–82) epitope in vitro from a synthetic polypeptide, and that inhibition of TPPII by AAF-CMK or by TPPII-specific small interfering RNA (siRNA) resulted in abrogation of epitope presentation in vivo. We propose that TPPII can work in combination with the proteasome or independent of the proteasome system to generate a subset of those epitopes that evade generation by the proteasome.
Results
HIV-1 Nef(73–83) is not generated by proteasomes
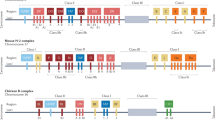
The HIV-1 Nef(73–82) epitope is an immunodominant CTL epitope recognized in 60% of infected patients in the context of both HLA-A3 and HLA-A11 MHC molecules16,21. To study the proteasome-mediated generation of this peptide, we digested a synthetic polypeptide derived from the Nef central immunodominant region (Nef amino acids 64–100) with 20S proteasomes in vitro. Independent of whether standard proteasomes or immunoproteasomes were used, we failed to generate the correct C terminus of the HLA-A3–restricted Nef(73–82) epitope. In agreement with previous observations16, we identified a dominant cleavage at residue Nef-Y81 that destroyed this major HLA-A3 epitope. Dominant cleavages were also observed at residues Nef-F68, Nef-A84 and Nef-L87 within the two flanking regions of the epitope (Fig. 1a). To exclude that the inability to generate the epitope was due to the use of 20S proteasome that is not the enzyme involved in antigen processing in vivo, we took advantage of the ornithine decarboxylase (ODC) system, which allows the ubiquitin-independent degradation of an ODC–fusion protein by 26S proteasomes in the presence of a chaperoning protein called antizyme22. Therefore, we constructed an ODC-Nef fusion protein containing the ovalbumin (OVA) epitope SIINFEKL as an internal control for antigen processing activity (Nef-ODC-OVA)22. Recombinant Nef-ODC-OVA was degraded in vitro by the 26S proteasome in the presence of recombinant antizyme, allowing the analysis of the degradation products of full-length Nef in a physiologically relevant system22. Although 26S proteasomes produced the OVA epitope, they failed to produce the Nef(73–82) epitope or a peptide carrying the correct C-terminal residue (Fig. 1b). The 26S proteasome degradation products also included the longer peptides Nef(69–84) and Nef(69–87), suggesting that these longer peptides were authentic Nef processing products. Thus, two different experimental approaches, which used either the 20S core complex in combination with a Nef(64–100) polypeptide or the 26S proteasomes in the context of an ODC-Nef fusion protein, resulted in the generation of an almost identical peptide pattern for the Nef region analyzed, but did not produce the desired epitope.

(a) Major cleavage products generated by 20S proteasomes from the Nef(64–100) polypeptide. (b) Major cleavage products generated by 26S proteasomes from the Nef-ODC-OVA fusion protein. (c) Major fragments generated by TPPII from the Nef(64–100) polypeptide. (d) Fragments generated by TPPII from a synthetic Nef(69–87) polypeptide. In both experiments, fragments directly flanking the epitope were identified. Dotted arrows mark fragments that are the result of a tripeptidyl trimming reaction. Solid arrows mark major cleavage sites. Dotted lines underline the location of the Nef(73–82) epitope.
Nef(73–82) presentation was insensitive to epoxomicin
To test Nef(73–82) presentation by professional APCs, we infected HLA-A3 human monocyte-derived DCs with the recombinant Vaccinia virus Vac-Nef, or as a control with Vac-RT, to induce the synthesis of HIV-1 Nef and reverse transcriptase, respectively. After overnight culture, Nef(73–82)-specific CD8+ T cell lines derived from HIV-infected patients were added to the DCs and tested for intracellular production of IFN-γ. Flow cytometric measurement of the expression of IFN-γ in CD8+ cells allows the assay of an effector response actually mediated by CD8+ T cells. In accordance with the in vitro processing data that showed the inability of proteasomes to generate the Nef epitope, HIV-Nef(73–82) presentation was not inhibited by the 20S and 26S proteasome inhibitors epoxomicin or MG132 (Fig. 2a,b). In contrast, recognition of the RT(476–484) epitope on Vac-RT infected, HLA-A2, HLA-A3 DCs by specific T cells was completely inhibited (Fig. 2c), as expected from its known proteasome-dependent generation23.

DCs infected with recombinant Vaccinia viruses or incubated with peptides were cultured overnight, then cocultured with CD8+ T cells that were tested for intracellular IFN-γ secretion; epoxomicin was added at 10 μM for 30 min before Vaccinia or peptide incubation, then diluted to 2 μM. (a,b) Nef(73–82)-specific T cells. (c) RT(476–484)-specific CD8+ T cells, to test the same DCs as in (a) in parallel. (d) TAP-negative (ST-EMO37) or TAP-positive (EBV-1) HLA-A3 B lymphoblastoid cells were infected or incubated with peptides overnight, then tested with Nef(73–82)-specific T cells. All experiments are representative of at least two experiments, except epoxomicin was tested at least six times at similar concentrations for Nef-specific responses. The average response percentages in the presence of epoxomicin compared with the noninhibited control were, for anti-Nef(73–82) responses, 115 ± 38% for Vac-Nef versus 115 ± 46% for Nef(73–82) (not significant).
These results raised the question of whether, in fact, HIV Nef(73–82) was generated by a cytosolic protease and therefore required transport from the cytosol into the ER by TAP for its presentation. We tested whether Nef(73–82) presentation after Vac-Nef infection was dependent on the presence of TAP. Surface presentation of Nef(73–82) was only observed in TAP-positive, EBV-transformed lymphoblastoid cells demonstrating the requirement for cytosolic Nef processing (Fig. 2d).
The observed TAP dependence of Nef(73–82) presentation indicated that processing of the peptide took place in the cytosol. This result, combined with the proteasome inhibitor independence of Nef(73–82) presentation, suggested that the proteasome system is either not rate-limiting or not responsible for the generation of this epitope.
In an attempt to identify the proteases involved in Nef-epitope generation, we tested various inhibitors of cytosolic proteases that were previously discussed to have a role in antigen presentation2,4,13,14. Treatment of DCs with ALLM (inhibits calpains)2, bestatin (inhibits various aminopeptidase including leucine amino peptidase and puromycin sensitive aminopeptidase)14 or E64 (inhibits cysteine proteases, calpains and the exo- and endoproteolytic activities of bleomycin hydrolase)14 revealed no effect on Nef(73–82) presentation (Fig. 3a,b). At the concentrations used in these experiments, the inhibitors were active in human DCs, as ALLM and E64 inhibited fluorogenic calpain substrate II digestion, and bestatin inhibited l-leucine p-nitroanilide substrate digestion in vitro with cell extracts of inhibitor-treated DCs (data not shown). Thus, the involvement of calpains2, bleomycin hydrolase14 or other cysteine proteases and leucine aminopeptidase13 was unlikely.

(a, b, c) DCs were tested as in Fig. 2a. (d) AAF-CMK (2 μM) was added for 30 min, washed and then added again (or not) for overnight culture. All experiments are representative of at least two experiments, except ALLM dose response curve and bestatin were tested only once, and AAF-CMK was tested six times at similar concentrations. The average response percentage in the presence of AAF-CMK compared with the noninhibited control was decreased for Vac-Nef compared with Nef(73–82): 32 ± 17% versus 94 ± 12% (P = 0.01).
Inhibition of HIV Nef(73–82) epitope presentation
The results of the experiments described above led us to speculate that the cytosolic protease TPPII19,24 may be responsible for Nef(73–82) epitope generation. Thus, DCs were treated with the known TPPII inhibitor Ala-Ala-Phe-chloromethylketone (AAF-CMK). Low concentrations (1 μM) of AAF-CMK inhibited the presentation of Nef(73–82) by 60%, and 20 μM of AAF-CMK abolished epitope presentation (Fig. 3c). AAF-CMK also inhibited Nef-epitope presentation in EBV-transformed lymphoblastoid cells, indicating that the role of TPPII was not restricted to DCs (data not shown).
To further substantiate the role of TPPII and exclude that of lysosomal TPPI, which is reversibly inhibited by AAF-CMK25, AAF-CMK was added to Vac-Nef–infected human DCs for 30 min and then washed away. Even this short period of AAF-CMK treatment irreversibly inhibited the presentation of the Nef(73–82) epitope (Fig. 3d). Thus, our data suggested that TPPII might be the rate-limiting cytosolic protease involved in generation of the Nef(73–82) epitope.
We hypothesized that if TPPII was responsible for Nef(73–82) epitope generation, then its endoproteolytic function would essentially be required for liberation of the Nef(73–82) epitope from a Nef(73–82)-containing polypeptide substrate.
Generation of the Nef(73–82) epitope by TPPII
To test the hypothesis that TPPII is responsible for Nef(73–82) epitope processing, we purified high-molecular-weight TPPII complexes to homogeneity from human erythrocytes. The identity of TPPII was verified by mass spectrometry, immunoblotting with TPPII antibody and its H-Ala-Ala-Phe-MCA hydrolyzing activity, which was completely inhibited by the TPPII-specific inhibitor butabindide26. The purity of the preparations was tested by SDS-PAGE (data not shown). To assay the antigen processing capacity of TPPII in in vitro processing experiments, we used the synthetic Nef(64–100) polypeptide containing the Nef(73–82) epitope as substrate (Fig. 1a). The analysis of the TPPII processing products showed that the enzyme generated the correct Nef(73–82) epitope with high efficiency (Fig. 1c). This TPPII-dependent epitope generation seemed to be the consequence of two endoproteolytic cleavages behind residues Nef-P72 and Nef-K82. Thus, in contrast to the 20S and 26S proteasomes, TPPII used the Nef K82-A83 peptide bond at the C terminus of the Nef epitope as a preferential endoproteolytic cleavage site. In addition, and allowing the liberation of the correct epitope from the polypeptide substrate, TPPII also generated the correct N terminus of the epitope by cleavage after residue Nef-P72 (Fig. 1c).
To study whether the length of the substrate influenced the efficiency of TPPII-dependent epitope production, we used a shorter polypeptide (Nef amino acids P69 through L87) that was shown to be a processing intermediate of the 20S and 26S proteasomes as substrate for TPPII processing (Fig. 1a,b). Again, TPPII used the C-terminal K82-A83 peptide bond as the major cleavage site (Fig. 1d). As evidenced by the identification of the N-terminal leaving fragment 69PVTP72, the generation of the correct N terminus was again the result of an endoproteolytic cleavage behind the Nef-P72 residue. The generation of the Nef(73–82) epitope by purified TPPII was impaired by butabindide, a specific inhibitor of TPPII, which, unlike AAF-CMK, did not permeate the cells.
TPPII siRNAs inhibit Nef(73–82) presentation
Both the inhibitor data and our in vitro processing data suggested that TPPII is most likely the rate-limiting cytosolic protease involved in generation of the Nef(73–82) epitope. To demonstrate that TPPII was indeed responsible for the generation of the Nef(73–82) epitope, Epstein Barr virus (EBV)-transformed lymphoblastoid cells were transfected with siRNAs for 48 h, then infected with Vac-Nef. Epitope recognition was inhibited up to 60% in a dose-dependent manner using the TPPII-specific siRNAs. In contrast, the scrambled control siRNAs had no effect on epitope presentation (Fig. 4). Our combined in vivo and in vitro data presented evidence that TPPII can be the rate-limiting enzyme for the generation of a MHC class I epitope with the correct C terminus, which could not be supplied by the proteasome.

EBV-1 cells were transfected with either TPPII- specific or scrambled siRNAs (scr.) for 48 h, infected or incubated with peptides overnight, then tested with Nef(73–82)-specific T cells. The experiment shown is representative of two.
Discussion
Based on the findings of proteasome cleavage characteristics, inhibitor studies, and in vitro and in vivo antigen processing experiments1,2,3,4,5, the proteasome seemed to be the only cellular enzyme with the capacity to generate the correct C termini and anchor residues of MHC class I ligands with the required efficiency. The involvement of other proteases in MHC class I antigen processing seemed to be restricted to the TAP-independent pathway6,27 or the trimming of N-terminally extended epitope precursor peptides11,12,14,28.
Previously it was found that loading of HLA-A3 binding peptides or maturation of HLA-A3, -A11 and -B35 molecules was insensitive to proteasome inhibition17. As lysine is not a preferred cleavage site for proteasomes, it was proposed that the frequent presence of lysine residues at the C terminus of HLA-A3 epitopes may interfere with proteasome function17,29. Here we showed that cytosolic TPPII is essential for the efficient generation of the immuno-dominant HLA-A3 and -A11–restricted HIV-1 Nef(73–82) epitope, which possesses a lysine as the C-terminal anchor residue.
From previous work in which proteasome activity was impaired by prolonged treatment of cells with the proteasome inhibitor lactacystin, it was proposed that TPPII may be able to perform basic proteasome functions such as, for example, the removal of misfolded proteins and may support cell survival19,30. The analysis of an OVA-derived polypeptide showed that TPPII possessed endo-proteolytic activity in addition to its aminopeptidase activity and that it degraded larger polypeptides19. In agreement with this, we found that TPPII generated an immuno-dominant HLA-A3 restricted CTL epitope by endo-proteolytic cleavages, indicating that TPPII can complement or even substitute proteasome function—also in the context of MHC class I antigen processing. It was suggested that the >2,000-kDa TPPII complex, which is composed of multiple 138-kDa subunits, may function as a compartmentalized enzyme like the proteasome31.
Although TPPII is a relatively abundant protein, there is very little known so far about its physiological function. Our analysis of HIV-Nef protein processing supports the concept that, under physiological conditions, TPPII functions downstream of an active ubiquitin-proteasome system, because TPPII was able to generate the HLA-A3 HIV-Nef(73–82) epitope from proteasomal Nef-protein processing intermediates. An efficient peptide supply is essential for MHC class I antigen presentation. Therefore, a cooperative action between the proteasome system and TPPII would guarantee that the infected cell is able to generate epitopes that cannot be efficiently generated by the proteasome system.
Our experiments, however, also demonstrated that inhibition of the proteasome system had no effect on TPPII-dependent production of the HLA-A3 HIV-Nef(73–82) epitope. This suggests that TPPII can also work in parallel to the proteasome system and can contribute to the MHC class I peptide pool independent of the proteasome. Thus, HIV-Nef(73–82) is most likely not the only epitope requiring TPPII activity. Such an independent role of TPPII may be of importance under conditions in which proteasome activity is impaired as a result of viral infections or metabolic stress. Thus, inhibition of proteasome activity seems to up-regulate TPPII activity30, and it has also been shown that viral proteins like HIV-TAT can directly interfere with proteasome function32.
These observations suggest that within the MHC class I antigen processing pathway, TPPII possesses a 'housekeeping' function under normal physiological conditions by taking up proteasomal processing intermediates, but that under conditions of physiological stress TPPII can also work independent of the proteasome and process protein with the help of chaperones or other yet to be defined protein factors.
Methods
Cell lines and cell culture.
DCs, Josk-M, T2 (TAP-deficient), T2-217 (T2 + immunoproteasome subunits33) and EBV-transformed lymphoblastoid cells were maintained in RPMI-1640 with 10% fetal calf serum (FCS). Nef(73–82)-specific CD8+ T cell lines were generated from HIV+ individuals from cohort studies established with approval of Cochin Hospital's ethics committee as described34. DCs were differentiated from elutriated monocytes cultured for 7 d in the presence of GM-CSF and IL-434.
Antigen presentation assays.
HLA-A2 and HLA-A3 DCs were infected with recombinant Copenhagen Vaccinia viruses encoding HIV-1 Lai nef or pol (VV.TG.1147 or 3167, Transgène, 5 plaque forming units (PFU) per cell, 5% FCS), or incubated with peptides overnight, then washed and assessed for viability. DCs were incubated with the CD8+ T cell lines and assayed for intracellular IFN-γ production in CD3+CD8+ lymphocytes by flow cytometry as described34.
Peptides, Nef-ODC fusion protein and inhibitors.
The peptides EEVGFPVTPQVPLRPMTYKAAVDLSHFLKEKGGLEGL (HIV Lai Nef amino acids 64–100), PVTPQVPLRPMTYKAAVDL (Nef amino acids 69–87) and QVPLRPMTYK (Nef amino acids 73–82) were synthesized using standard Fmoc methodology on an Applied Biosystems (Norwalk, CT) 433A automated synthesizer at >90% purity by the peptide synthesis group of the Institute of Biochemistry-Charité. The peptide ILKEPVHGV (RT 476–484) was obtained from Neosystem (Strasbourg, France). Expression and purification of Nef-ODC-OVA and purification of the maltose binding protein-antizyme (MBP-AZ) were performed as described22. Protease inhibitors were epoxomicin (added at 10 μM 30 min before infection, then diluted to 2 μM; Alexis, Grünberg, Germany), and MG 132, bestatin, E64, AAF-CMK (Sigma, Taufkirchen, Germany), ALLM, LLnL (Calbiochem, Schwalbach, Germany) and butabindide26 (added 30 min after vaccinia infection). The fluorogenic calpain substrate II (Calbiochem) and the L-leucine p-nitroanilide substrate (Sigma) were used as controls to check for enzyme inhibitor activities in human DCs.
Proteasome isolation.
The 20S and 26S proteasomes were essentially purified as described22,35. The 20S constitutive proteasomes were isolated from Josk-M and T2 (TAP-deficient) cells, and 20S immunoproteasomes were isolated from T2-217 (T2 + immunoproteasome subunits33) cells and from Josk-M cells stimulated for 72 h with 200 U/ml IFN-γ. The 26S proteasomes were purified from human red blood cells.
TPPII purification.
TPPII was purified from human erythrocytes at 4 °C. The 100,000g supernatant obtained from washed and lysed erythrocytes was mixed with 100 g DEAE-Cellulose SERVACEL (SERVA, Heidelberg, Germany) in TEAD buffer (20 mM Tris/HCL (pH 7.5), 1 mM EDTA, 1 mM NaN3, 1 mM Dithioerythrit). After washing with TEAD, bound proteins were eluted with 500 mM NaCl in TEAD, then further fractionated by ammonium sulfate. Proteins precipitating between 35% and 70% saturation were pelleted at 15,000g, resuspended, dialyzed against TEAD and applied to a DEAE-Sephacel-column in 50 mM NaCl, TEAD. Proteins were eluted with a linear gradient of 50–500 mM NaCl, TEAD. Fractions containing H-AAF-MCA hydrolyzing activity were pooled. Residual proteasomes were removed by affinity chromatography with monoclonal antibody mcp2136. Unbound TPPII exhibiting H-AAF-MCA hydrolyzing activity inhibited by H-AAF-CMK was further purified by successive chromatography on MonoQ, arginine-Sepharose 4B and Superose 6B. All columns were equilibrated in 20 mM HEPES (pH 7.2), 15% glycerol and 1 mM ATP. TPPII was eluted from Mono Q and arginine-Sepharose columns with linear increasing gradients (0–400 mM NaCl in HEPES, ATP, glycerol). The purity of TPPII was checked by SDS-PAGE combined with immunoblot analysis using polyclonal chicken anti-(human TPPII) Ig (Immunsystem, Uppsala, Sweden). In some preparations minor amounts of spectrin (major erythrocyte component) and β-actin copurified as judged by mass spectrometric sequencing (MS/MS) analysis. TPPII activity was confirmed by digestion of fluorogenic peptide substrate and its complete inhibition by butabindide. TPPII activity was insensitive to the proteasome inhibitor N-acetyl-l-leucinyl-l-leucinal-l-norleucinal (LLnL).
Peptide digestion and mass spectrometry.
Nef(64–100) polypeptide (20 μg) and 20S proteasomes (2 μg) were incubated in 300 μl assay buffer (20 mM HEPES/KOH, pH 7.8, 2 mM MgAc2, 1 mM dithiothreitol) at 37 °C for different times. Nef-ODC-OVA fusion protein was incubated with 26S proteasome22. Nef(64–100) and Nef(69–87) (10 μg) were incubated with 100 ng or 1 μg TPPII in 50 μl assay buffer and incubated for 3, 24 and 48 h at 37 °C in the presence of the proteasome inhibitor LLnL. TPPII-dependent processing was sensitive to butabindide. Reversed-phase chromatography and mass spectrometric (MS) analyses performed online with an ion trap mass spectrometer (LCQ, Thermo-Finnigan, Engelsbach, Germany) equipped with an electrospray ion source were performed as described35. Peptides were identified by tandem mass spectrometry experiments. Cleavage products from the Nef-ODC-OVA fusion protein were analyzed as described22.
siRNAs and electroporation.
We synthesized 21-nucleotide interfering RNA duplexes with two 3′ end overhang dT nucleotides in the antisense strand. The sequences of the antisense strands of the siRNAs targeting TPPII were 5′- GUGGCGAUGUGAAUACUGCdTdT-3′, and the control scrambled oligoribonucleotide scrambled-siRNA was 5′-UGUAUAGGUGUGGGCACACdTdT-3′ (Eurogentec, Seraing, Belgium). TPPII expression was decreased in transfected HeLa cells in fluorogenic substrate digestion assays and in immunoblots. Transfection used an ECM 830 square wave electroporation system (BTX, San Diego, CA). Briefly, 4 × 105 EBV-transformed lymphoblastic cells were washed twice in PBS and placed in 4-mm gap cuvettes in the presence of oligonucleotides, subjected to 5 cycles of 20 V for 10 ms separated by 500-ms gaps in electroporation buffer (120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4/KH2PO4, 25 mM HEPES, 2 mM EGTA, 5 mM MgCl2, 50 mM glutathione, 2 mM ATP, pH 7.6). Cells were washed again and transferred to culture medium for 48 h before incubating with Vaccinia viruses and performing antigen presentation assays.
References
Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2, 179–187 (2001).
Rock, K.L. et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771 (1994).
York, I.A., Goldberg, A.L., Mo, X.Y. & Rock, K.L. Proteolysis and class I major histocompatibility complex antigen presentation. Immunol. Rev. 172, 49–66 (1999).
Niedermann, G. et al. The specificity of proteasomes: impact on MHC class I processing and presentation of antigens. Immunol. Rev. 172, 29–48 (1999).
Tanaka, K., Tanahashi, N., Tsurumi, C., Yokota, K.Y. & Shimbara, N. Proteasomes and antigen processing. Adv. Immunol. 64, 1–38 (1997).
Yewdell, J.W. & Bennink, J.R. Cut and trim: generating MHC class I peptide ligands. Curr. Opin. Immunol. 13, 13–18 (2001).
Knuehl, C. et al. The murine cytomegalovirus pp89 immunodominant H-2Ld epitope is generated and translocated into the endoplasmic reticulum as an 11-mer precursor peptide. J. Immunol. 167, 1515–1521 (2001).
Mo, X.Y., Cascio, P., Lemerise, K., Goldberg, A.L. & Rock, K. Distinct proteolytic processes generate the C and N termini of MHC class I-binding peptides. J. Immunol. 163, 5851–5859 (1999).
Serwold, T., Gaw, S. & Shastri, N. ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat. Immunol. 2, 644–651 (2001).
Fruci, D., Niedermann, G., Butler, R.H. & van Endert, P.M. Efficient MHC class I-independent amino-terminal trimming of epitope precursor peptides in the endoplasmic reticulum. Immunity 15, 467–476 (2001).
Saric, T. et al. An IFN-γ-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat. Immunol. 3, 1169–1176 (2002).
Serwold, T., Gonzalez, F., Kim, J., Jacob, R. & Shastri, N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature 419, 480–483 (2002).
Beninga, J., Rock, K.L. & Goldberg, A.L. Interferon-γ can stimulate post-proteasomal trimming of the N terminus of an antigenic peptide by inducing leucine aminopeptidase. J. Biol. Chem. 273, 18734–18742 (1998).
Stoltze, L. et al. Two new proteases in the MHC class I processing pathway. Nat. Immunol. 1, 413–418 (2000).
Lucchiari-Hartz, M. et al. Cytotoxic T lymphocyte epitopes of HIV-1 Nef: Generation of multiple definitive major histocompatibility complex class I ligands by proteasomes. J. Exp. Med. 191, 239–252 (2000).
Choppin, J. et al. Characteristics of HIV-1 Nef regions containing multiple CD8+ T cell epitopes: wealth of HLA-binding motifs and sensitivity to proteasome degradation. J. Immunol. 166, 6164–6169 (2001).
Benham, A.M., Gromme, M. & Neefjes, J. Allelic differences in the relationship between proteasome activity and MHC class I peptide loading. J. Immunol. 161, 83–89 (1998).
Tomkinson, B., Wernstedt, C., Hellman, U. & Zetterqvist, O. Active site of tripeptidyl peptidase II from human erythrocytes is of the subtilisin type. Proc. Natl. Acad. Sci. USA 84, 7508–7512 (1987).
Geier, E. et al. A giant protease with potential to substitute for some functions of the proteasome. Science 283, 978–981 (1999).
Levy, F. et al. The final N-terminal trimming of a subaminoterminal proline-containing HLA class I-restricted antigenic peptide in the cytosol is mediated by two peptidases. J. Immunol. 169, 4161–4171 (2002).
Culmann-Penciolelli, B. et al. MHC class I multirestriction in the central region of the VIH-1 Nef protein explained at the molecular level. J. Virol. 68, 7336–7342 (1994).
Ben-Shahar, S. et al. 26 S proteasome-mediated production of an authentic major histocompatibility class I-restricted epitope from an intact protein substrate. J. Biol. Chem. 274, 21963–21972 (1999).
Sewell, A.K. et al. IFN-γ exposes a cryptic cytotoxic T lymphocyte epitope in HIV-1 reverse transcriptase. J. Immunol. 162, 7075–7079 (1999).
Balow, R.M., Tomkinson, B., Ragnarsson, U. & Zetterqvist, O. Purification, substrate specificity, and classification of tripeptidyl peptidase II. J. Biol. Chem. 261, 2409–2417 (1986).
Hilbi, H., Puro, R.J. & Zychlinsky, A. Tripeptidyl peptidase II promotes maturation of caspase-1 in Shigella flexneri-induced macrophage apoptosis. Infect. Immun. 68, 5502–5508 (2000).
Rose, C. et al. Characterization and inhibition of a cholecystokinin-inactivating serine peptidase. Nature 380, 403–409 (1996).
Gil-Torregrosa, B.C., Raul Castano, A. & Del Val, M. Major histocompatibility complex class I viral antigen processing in the secretory pathway defined by the trans-Golgi network protease furin. J. Exp. Med. 188, 1105–1116 (1998).
Komlosh, A. et al. A role for a novel luminal endoplasmic reticulum aminopeptidase in final trimming of 26 S proteasome-generated major histocompatibility complex class I antigenic peptides. J. Biol. Chem. 276, 30050–30056 (2001).
Falk, K. et al. Peptide motifs of HLA-A1, -A11, -A31, and -A33 molecules. Immunogenet. 40, 238–241 (1994).
Wang, E.W. et al. Integration of the ubiquitin-proteasome pathway with a cytosolic oligopeptidase activity. Proc. Natl. Acad. Sci. USA 97, 9990–9995 (2000).
Tomkinson, B. Tripeptidyl peptidases: enzymes that count. Trends Biochem. Sci. 24, 355–359 (1999).
Huang, X. et al. The RTP site shared by the HIV-1 Tat protein and the 11S regulator subunit α is crucial for their effects on proteasome function including antigen processing. J. Mol. Biol. 323, 771–782 (2002).
Sijts, A.J. et al. Efficient generation of a hepatitis B virus cytotoxic T lymphocyte epitope requires the structural features of immunoproteasomes. J. Exp. Med. 191, 503–514 (2000).
Andrieu, M. et al. Two HIV vaccinal lipopeptides follow different cross-presentation pathways in human dendritic cells. J. Virol. 77, 1564–1570 (2003).
Groettrup, M. et al. The interferon-γ-inducible 11S regulator and the LMP2/LMP7 subunits govern the peptide production by the 20S proteasome in vitro. J. Biol. Chem. 270, 23808–23815 (1995).
Hendil, K.B. & Uerkvitz, W. The human multicatalytic proteinase: affinity purification using a monoclonal antibody. J. Biochem. Biophys. Methods 22, 159–165 (1991).
de la Salle, H. et al. Homozygous human TAP peptide transporter mutation in HLA class I deficiency. Science 265, 237–241 (1994).
Acknowledgements
We thank B. Dahlmann for helpful advice in the purification of TPPII; D. Hanau and R. Drillien for monocytes and TAP-negative EBV-transformed lymphoblastoid; C.R. Ganellin for butabindide; M.P. Kieny for recombinant vaccinia viruses; M. Lichtner and F. Mengoni for calpain test reagents; A. Sijts and U. Kuckelkorn for helpful discussions; and T. Meyer for support. L.W. was supported by a grant of the Charité. U.S. was partially supported by the Deutsche Forschungsgemeinschaft. The work was supported by the Deutsche Forschungsgemeinschaft (P.-M.K. and H.S.), INSERM (A.H. and C.M.), the European Community (C.M.), the Agence Nationale de Recherche sur le SIDA (ANRS), Ensemble Contre le SIDA (Sidaction) and the Research Career Development Award by the Israel Cancer Research Foundation (Y.R.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Seifert, U., Marañón, C., Shmueli, A. et al. An essential role for tripeptidyl peptidase in the generation of an MHC class I epitope. Nat Immunol 4, 375–379 (2003). https://doi.org/10.1038/ni905
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni905
This article is cited by
-
Role of peptide processing predictions in T cell epitope identification: contribution of different prediction programs
Immunogenetics (2015)
-
Census of cytosolic aminopeptidase activity reveals two novel cytosolic aminopeptidases
Medical Microbiology and Immunology (2012)
-
Insights into the processing of MHC class I ligands gained from the study of human tumor epitopes
Cellular and Molecular Life Sciences (2011)
-
Post-proteasomal and proteasome-independent generation of MHC class I ligands
Cellular and Molecular Life Sciences (2011)
-
Antigen processing by nardilysin and thimet oligopeptidase generates cytotoxic T cell epitopes
Nature Immunology (2011)
