
Abstract
Charge transfer (CT) states at a donor–acceptor heterojunction have a key role in the charge photogeneration process of organic solar cells, however, the mechanism by which these states dissociate efficiently into free carriers remains unclear. Here we explore the nature of these states in small molecule–fullerene bulk heterojunction photovoltaics with varying fullerene fraction and find that the CT energy scales with dielectric constant at high fullerene loading but that there is a threshold C60 crystallite size of ~4 nm below which the spatial extent of these states is reduced. Electroabsorption measurements indicate an increase in CT polarizability when C60 crystallite size exceeds this threshold, and that this change is correlated with increased charge separation yield supported by CT photoluminescence transients. These results support a model of charge separation via delocalized CT states independent of excess heterojunction offset driving energy and indicate that local fullerene crystallinity is critical to the charge separation process.
Similar content being viewed by others
Introduction
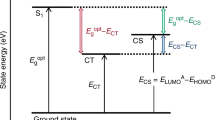
Charge transfer (CT) states at a donor–acceptor heterojunction (DA HJ) are increasingly recognized as key in determining both the practical and thermodynamic limiting efficiency of organic photovoltaic cells1,2,3,4,5,6. These states result from photoinduced CT at the DA HJ and consist of a Coulombically correlated hole and electron perceived to reside on adjacent (or closely neighbouring) donor and acceptor molecules, respectively. Although the important role of CT states in determining photocurrent generation and open-circuit voltage has been established7,8,9, the means by which they overcome an estimated binding energy ~10 kbT to achieve efficient charge separation at room temperature has been the subject of debate1,2.
The classical Coulomb estimate of relaxed CT-binding energy, EB=q2/4πεrCT, depends inversely on dielectric constant, ε, and the length rCT, which reflects some average electron and hole separation indicative of the CT spatial extent2. It is unclear, however, what the relevant frequency range of ε(ω) is or even how much influence such a macroscopically averaged quantity should have on states existing between adjacent molecules. An alternative perspective has emerged from recent spectroscopic investigations suggesting that, at early times following CT (<1 ps), delocalized CT states can be populated that efficiently sample and decay into free charge carriers before ever localizing in the Coulomb well3,4.
Here, we explore the dependence of CT energy, ECT, on background dielectric constant directly via electroluminescence (EL), photoluminescence (PL) and absorption by varying the blend ratio of small molecule–bulk heterojunction (BHJ) organic solar cells based on the donor N,N′-bis(1-naphthyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine (NPD) and the acceptor C60. Consistent with previous observations, we observe a red-shift of ECT with increasing C60 fraction10,11,12,13,14, but find that modelling based on the accompanying change in dielectric constant via the solid-state solvation effect (SSSE)15 can only explain the data at high (>50%) C60 loading. We find that ECT at low fullerene concentration is higher than expected from the change in dielectric constant and attribute this discrepancy to increased localization of the electron component of the CT state because of a reduction in average C60 crystallite size below ~4 nm. Using electroabsorption (EA) spectroscopy16, we observe a substantial increase in CT state polarizability beyond this threshold crystallite size indicative of increasing delocalization, and find that this leads to rapid decay in the CT PL transient attributed to efficient long-range charge separation. These results point to the importance of local crystallinity in enabling delocalization of at least one component (e− or h+) of the CT state as well as the need to include multiple donor and acceptor molecules in theoretical modelling to accurately capture extended CT states relevant in the charge separation process.
Results
CT energy dependence on dielectric constant
BHJ devices were fabricated with active layer composition ranging from 20 to 90% C60 according to the relative co-deposition ratios 4:1, 2:1, 1:1, 1:4 and 1:9 NPD to C60. Capacitance–voltage measurements were subsequently carried out at a frequency of 100 kHz with devices depleted in reverse bias (−2 V) to extract the low frequency relative dielectric constant (εstatic) of each BHJ layer. These data are plotted in Fig. 1a together with the optical frequency dielectric constants (εoptical) fit from spectroscopic ellipsometry at an energy E=1.26 eV near the peak of the CT state emission. There is little difference between εstatic and εoptical and both vary approximately linearly with C60 fraction, consistent with expectations of an effective medium description.

(a) Static (100 kHz) and optical frequency dielectric constants measured as a function of C60 volume fraction in the blend. The molecular structure of N,N′-bis(1-naphthyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine (NPD) is shown in the inset. Error bars for εoptical reflect standard error in the ellipsometry fits, whereas those for εstatic indicate standard deviations obtained over multiple device measurements. (b) Dark current–voltage characteristics of bulk heterojunction (BHJ) devices with different NPD:C60 ratios. Inset: short-circuit currents (Jsc) and internal quantum efficiencies (IQE, λ=450 nm) of the different BHJ devices.
The dark current density–voltage (J-V) characteristics of each device are shown in Fig. 1b together with their short-circuit currents (Jsc) under AM1.5G illumination and internal quantum efficiency (IQE) measured at λ=450 nm (inset). Similar to many polymer/fullerene11 and evaporated17 BHJs, we find that overall efficiency is maximized at high C60 concentration because of a substantial drop in Jsc with increasing NPD fraction, which is primarily due to decreasing IQE as opposed to reduced C60 absorption (NPD absorbs little sunlight with a 3.1-eV optical gap). The functional form of the J-V characteristic is qualitatively maintained with increasing NPD fraction up to 2:1 NPD:C60 loading, indicating that current is controlled by the same DA HJ generation/recombination processes9, albeit with different strengths expected from changing carrier mobilities and recombination rates18.
Figure 2a shows the EL spectra measured under forward bias at current densities of 75 mA cm−2, which exhibit a peak assigned to an NPD-C60 CT state that red-shifts from approximately 1.3 to 1.2 eV with increasing C60 concentration. This CT state is the sole EL component in the 1:9, 1:4 and planar HJ device spectra, whereas some NPD and C60 excitonic emission (at peak energies E=2.9 eV and 1.67 eV, respectively; see Supplementary Fig. 1) is observed in the 1:1, 2:1 and 4:1 spectra as higher bias (up to 4 V) was needed to achieve a current density with sufficient signal to noise in the CT emission. No measurable change in CT emission energy was observed as a function of bias (see Supplementary Fig. 2), indicating that filling within the disorder-induced CT density of states is negligible in these measurements.

(a) Electroluminescence (EL) spectra indicating a shift in charge transfer (CT) emission in cells with varying N,N′-bis(1-naphthyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine (NPD):C60 ratio. The spectra are normalized and shifted vertically for clarity. (b) EL peak energies extracted from fitting and plotted as a function of static dielectric constant. The red dashed line indicates the prediction of solid-state solvation theory (SSSE) described in the text. The inset shows the same CT peak energies plotted versus inverse dielectric constant. Error bars indicate standard error obtained in the EL peak fitting of a. NA, not applicable; PHJ, planar heterojunction.
The CT red-shift with increasing fullerene concentration has previously been attributed to the accompanying increase in dielectric constant (c.f. Fig. 1a)10,11. To test this hypothesis, the CT peak energies were extracted from Gaussian multipeak fitting and plotted in Fig. 2b against εstatic and its inverse (inset). A nearly identical dielectric constant dependence was observed for the CT peak in PL as well (see Supplementary Fig. 3).
The energy shift can be partially explained via the SSSE, which successfully describes the emission red-shift of other polar excited states with increasing dielectric constant in organic thin films15. Here, the energy shift ΔEem=−AΛstatic−BxΛoptical accounts for screening by the induced local electric field and involves contributions from both the static and optical frequency dielectric constants according to the local field factors Λ=2(ε−1)/(2ε+1) and constants A and B that depend on the ground and excited state dipole moments involved in the optical transition.
Comparing this relationship to the data of Fig. 2, we find that it can account for the emission energy shift of the 1:9, 1:4 and 1:1 BHJs, but that it fails at lower C60 fraction. The SSSE fit indicated in Fig. 2b is not unique because of the similarity between εstatic and εoptical (that is, A and B are correlated); however, the key point here is that this description cannot, even in principal, describe the full data range—there is simply more curvature in the emission energy shift than the SSSE model is able to predict. Alternatively, Veldman et al.11 explained shifts in polymer/fullerene CT emission according to an electrostatic expression for the Gibbs free energy for photoinduced electron transfer, which carries the Coulomb electrostatic scaling ΔEem  1/εrCT. However, as evident in the inset of Fig. 2b, the emission observed here does not shift linearly with inverse dielectric constant, suggesting that the CT spatial extent (that is, rCT) may be changing at low C60 fraction.
1/εrCT. However, as evident in the inset of Fig. 2b, the emission observed here does not shift linearly with inverse dielectric constant, suggesting that the CT spatial extent (that is, rCT) may be changing at low C60 fraction.
A change in CT energy is also inferred in absorption through sensitive measurement of the external quantum efficiency (EQE). These data are shown in Fig. 3a and feature an absorption tail below the NPD and C60 optical gaps (see Supplementary Fig. 4) that clearly red-shifts with increasing C60 concentration. Similar to previous analyses of polymer/fullerene blends7,19, we assign this feature to absorption from the CT state and find that its low-energy tail can be described by the relationship:

(a) External quantum efficiency (EQE) data for devices with varying N,N′-bis(1-naphthyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine (NPD):C60 ratio, normalized at 1.7 eV for comparison. Green lines indicate fits to Equation 1 in the text and the inset displays the energy shift occurring along the dashed line at constant EQE=10−2. Error bars in the inset reflect the standard deviations estimated from EQE measurements on multiple devices. (b) Glancing incidence X-ray diffraction data for each of the bulk heterojunction films along with a neat C60 film.

which is characteristic of a transition based on Marcus photoinduced electron transfer7,20. Here, λr is the reorganization energy and the constant C0 includes the transition dipole moment. Fits to this expression (green lines) yield CT absorption maxima with an average of ECT+λr=1.85 eV, although the imprecision expected in fitting only half of the inverted parabola introduces correlation between the vertex position and broadening parameters that precludes us from establishing a meaningful trend among the different NPD:C60 blend ratios. Instead, we quantify the shift by plotting the energy change at a constant EQE value identified by the dashed line as shown in the inset21. Although it is difficult to distinguish whether this trend is due to a shift or broadening of the CT transition (more insight is obtained from EA below), the ~70 meV overall magnitude of the change is at least qualitatively consistent with that observed in the EL and PL measurements.
In addition to dielectric constant, the degree of C60 crystallinity in the BHJs reduces with decreasing C60 concentration as evident from glancing incidence X-ray diffraction data shown in Fig. 3b and well-documented in polymer-fullerene systems13,14,22. Application of the Scherrer equation to Gaussian fits of the C60 reflection at ~11° yields average crystallite sizes of 8.1, 6.5, 5.6, 4.2 and 3.8 nm for the pure C60, 1:9, 1:4, 1:1 and 2:1 BHJ films, respectively. No peak could be extracted from either the 4:1 BHJ or neat NPD films as they are highly disordered and most likely amorphous.
Based on these observations, we hypothesize that the increase in CT emission energy at low C60 concentration, also noted by Piersimoni et al.13 and Muller et al.14 in polymer-fullerene blends, is due to confinement of the wavefunction as C60 crystallite size becomes comparable to the natural spatial extent of the CT state (that is, that for large crystallites where CT volume is not influenced by the crystallite boundaries), similar to the manner in which exciton energy increases with decreasing inorganic semiconductor quantum dot size23. In this scenario, the electron component of the CT wavefunction in 1:9 and 1:4 BHJs is delocalized over two or three C60 molecules within a crystallite, whereas the hole remains localized on an adjacent NPD molecule. This estimate is based on the 1.42 nm C60 FCC lattice spacing, which corresponds to approximately three C60 molecules in any given dimension for the average 4.2 nm crystallite in the threshold 1:1 BHJ; further reduction in crystallite size increasingly confines the electron and raises the overall energy of the CT state.
CT state EA
We have examined this possibility further by conducting EA measurements, depleting each cell at −1 V bias and adding a 300-mVrms dither at a frequency of 1 kHz to create a uniform applied field F=FDC+FACcos(ωt) throughout the device. The Stark shift in transition energy because of this field is obtained from second-order perturbation theory as ΔE=−Δμ·F−[1/2] Δ
Δ
 F2, where Δμ is the difference between ground and excited state dipole moments and Δ
F2, where Δμ is the difference between ground and excited state dipole moments and Δ is the average change between the ground and excited state polarizability tensors16,24. Taylor expanding the change in absorption coefficient due to this energy shift as Δα=α′ΔE+[α″/2](ΔE)2 and averaging over randomly oriented molecules/crystallites, we find the first-order (rms) response at the modulation frequency16,24:
is the average change between the ground and excited state polarizability tensors16,24. Taylor expanding the change in absorption coefficient due to this energy shift as Δα=α′ΔE+[α″/2](ΔE)2 and averaging over randomly oriented molecules/crystallites, we find the first-order (rms) response at the modulation frequency16,24:

The differential transmission is measured with a lock-in amplifier and is related to the change in absorption coefficient, dΔα=−ΔT/T, where the optical path length d is double the active layer thickness as light is collected in reflection off the Ag cathode. Equation 2 predicts that the EA spectrum will follow the negative first derivative of the absorption coefficient (−α′) if a polarizability shift dominates, as in the case of, for example, delocalized J-aggregate excitons25, whereas it will follow the second derivative (α″) if the change in dipole moment is dominant as expected for CT states.
Figure 4 shows the EA spectra obtained for the 1:4 and 4:1 devices along with their respective linear absorption coefficients. At energies >2.2 eV, the 1:4 EA spectrum in Fig. 4b is dominated by a second derivative lineshape arising from known CT transitions in bulk C60 that yield a dipole moment of Δμ=24 D (refs 24, 26). The 4:1 spectrum in Fig. 4c is correspondingly dominated by the second derivative NPD feature at 3 eV, although the magnitude of the dipole moment Δμ=5 D is much smaller because of the Frenkel nature of the NPD exciton. The C60 feature in this case, which is below the derivative resolution of the linear absorption, is blue-shifted relative to the 1:4 BHJ, likely due to a combination of smaller crystallite size and lower dielectric constant.

(a) Linear absorption coefficients measured for 1:4 and 4:1 N,N′-bis(1-naphthyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine (NPD):C60 films. (b) EA spectra collected for the 1:4 device and (c) for the 4:1 device. Blue lines show second derivatives (α'') of the respective absorption coefficients scaled in magnitude to the EA data.
In both spectra, we observe additional features below 2.2 eV that are not evident in the EA of either pure NPD or C60 devices (see Supplementary Fig. 5). The most evident is located at ~1.85 eV, which corresponds closely with the CT absorption maximum identified through EQE fitting in Fig. 3a. The fact that these devices are depleted and this feature maintains the same modulation frequency dependence (up to the 250 kHz upper limit of the lock-in) as the higher energy excitonic EA is evidence against charge modulated (that is, polaron) absorption and thus we assign the 1.85 eV feature to CT EA.
Figure 5 provides a closer examination of the CT EA, where we observe a systematic change in moving from a dip in the 4:1 BHJ to a first derivative shape in the 1:9 device. Reproducing the CT absorption lineshape (αCT) fit from the EQE in Fig. 5a, we see that the dip corresponds to the second derivative, α″CT, expected for a typical CT transition in the 4:1 blend, but that this steadily evolves to include a growing contribution of first derivative lineshape (−α′CT) in the 1:9 blend, indicating a substantial increase in polarizability. This is demonstrated qualitatively by the green lines in Fig. 5b that comprise weighted superpositions of −α′CT and α″CT, where the contribution from −α′CT grows from negligible in the 4:1 to dominant in the 1:9 BHJ. For the sake of transparency, no attempt is made to incorporate the increasing high energy C60-related background in the 1:4 and 1:9 mixtures.

(a) CT absorption lineshape (αCT) along with its first (α'CT) and second (α''CT) derivatives estimated according to Equation 1 of the text and the green line fits in Fig. 3a. (b) Low-energy electroabsorption spectra for each NPD:C60 BHJ ratio, showing a systematic increase in first derivative lineshape component with increasing C60 fraction. BHJ, bulk heterojunction; NPD, N,N′-bis(1-naphthyl)-N,N′-diphenyl-1,1′-biphenyl-4,4′-diamine.
Discussion
The significant increase in polarizability suggests that the CT state becomes increasingly delocalized and occupies a greater volume at high C60 loading. This is both intuitively evident and more rigorously from the matrix elements that make up the polarizability, pCT~ /(En−ECT), as these reflect the ease with which neighbouring states, |ψn, mix with the CT state, |ψCT (ref. 27). Quantitative extraction of the CT dipole moment and its growing polarizability is not possible directly from the linear absorption data in Fig. 4a as first- and second-derivative components of the weak CT absorption cannot be resolved from background noise fluctuations (for example, small differences in reflection and/or scattering). Employing a more sensitive, non-transmission-based absorption technique such as photothermal deflection spectroscopy could resolve this issue. Nevertheless, based on the known slope of αCT according to Equation 1 and its approximate absorption coefficient relative to C60 from EQE, we estimate μCT>20 D in the 4:1 blend and
/(En−ECT), as these reflect the ease with which neighbouring states, |ψn, mix with the CT state, |ψCT (ref. 27). Quantitative extraction of the CT dipole moment and its growing polarizability is not possible directly from the linear absorption data in Fig. 4a as first- and second-derivative components of the weak CT absorption cannot be resolved from background noise fluctuations (for example, small differences in reflection and/or scattering). Employing a more sensitive, non-transmission-based absorption technique such as photothermal deflection spectroscopy could resolve this issue. Nevertheless, based on the known slope of αCT according to Equation 1 and its approximate absorption coefficient relative to C60 from EQE, we estimate μCT>20 D in the 4:1 blend and  CT~2,600 Å3 in the 1:4 and 1:9 BHJs, which is roughly three times larger than the polarizability found for the CT-related transitions at 2.4 eV in pure C60 (ref. 24).
CT~2,600 Å3 in the 1:4 and 1:9 BHJs, which is roughly three times larger than the polarizability found for the CT-related transitions at 2.4 eV in pure C60 (ref. 24).
PL transient data of the CT emission shown in Fig 6 indicate that delocalization is accompanied by a significant difference in decay dynamics. Whereas the localized CT state in the 4:1 blend exhibits an approximately monoexponential decay with lifetime τ=290±10 ps, the 1:4 blend displays a rapid initial decay followed by the same lifetime. This rapid initial decay reflects dissociation of delocalized CTs, whereas the low-level τ=290 ps tail is probably due to decay of localized CTs in the fraction of smaller (sub-4 nm) C60 crystallites within the BHJ ensemble.

Transient CT photoluminescence in the energy range 1.39–1.45 eV for bulk heterojunctions with high (1:4) and low (4:1) C60 fraction.
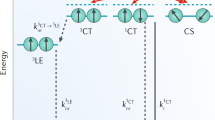
Taken together, our observations point to an important role of local fullerene order (and also potentially of the donor in other material systems) beyond a threshold crystallite size of ~4 nm in enabling CT delocalization that is key to efficient long-range charge separation. Moreover, in the presence of sufficiently sized C60 crystallites, CT delocalization is not dependent on excess driving energy from the initial CT reaction as it is observed here under resonant EA excitation28,29. The impact of fullerene nanocrystallinity on charge separation has been noted previously, where it was attributed to high local electron mobility within the traditional Onsager framework11,30 as well as to shifts in electron affinity of the fullerene crystallites31. In contrast, our results here point to the role of delocalization, analogous to recent findings for organic–inorganic HJs32, and are consistent with a growing number of recent reports in which charge separation is driven by accessing band-like CT states that subsequently decay on a sub-50 ps timescale (an upper limit based on the resolution of our PL transient data) into separated, free charge carriers3,4,29,33.
In summary, these results support a model of delocalized CT state dissociation and show that this process depends critically on nanoscale fullerene crystallinity. It is expected that this conclusion applies more generally to the donor as well as other acceptors (nanocrystallinity of both D and A is not required), although this will depend on the degree of intermolecular electronic coupling within the crystallites, the strength of which may be one reason for the unique success of fullerenes as organic photovoltaic acceptors. Looking forward, it is important for theoretical efforts to include many molecules in modelling a DA HJ to accurately capture the role of transient extended states in the photogeneration process and support the design of new donor and acceptor material combinations with lower energy loss and higher open-circuit voltage.
Methods
Device fabrication
BHJ devices were fabricated by thermal evaporation onto pre-patterned indium tin oxide (ITO) contacts according to the structure: glass/ITO (100 nm)/MoO3 (5 nm)/NPD:C60 (40 nm)/bathocuproine (10 nm)/Ag (100 nm). The active layer composition was varied from 20 to 90% C60 according to the relative co-deposition rates of NPD to C60 in the ratios 4:1, 2:1, 1:1, 1:4 and 1:9. Films for PL were deposited separately on quartz followed by a 100-nm Ag layer to minimize substrate autofluorescence and maintain a comparable optical environment as in the electrical devices. All devices and films were packaged in a glove box using ultraviolet-curable epoxy and a glass cover to avoid atmospheric degradation. Device performance metrics measured under AM1.5G simulated 1 sun illumination are summarized in Supplementary Fig. 6.
Ellipsometry and X-ray diffraction
Spectroscopic measurements on Si/SiO2 (100 nm)/organic (NPD, C60 or NPD:C60; 50 nm) layers were conducted using a variable angle spectroscopic ellipsometer (Ges5, Sopralab); ellipsometry refractive index dispersions are shown in Supplementary Fig. 7. On the same films, glancing incidence X-ray diffraction measurements were performed on a PANalytical X’Pert Pro Materials Research Diffractometer (PANalytical) using Cu Kα radiation. An integration time of 5 s per 2θ of 0.01° was used for scanning 2θ, with a fixed incident angle of 0.2°.
Luminescence and EA
EQE data were obtained using a standard monochromator and lock-in amplifier setup with a laser-driven Xe lamp source and a chopping frequency of 500 Hz. IQE was calculated from the balance of reflected and transmitted light measured in the same experiment (that is, IQE=EQE/(1−R−T)). EA data were acquired using the same system with light incident at an angle of ~15° through the ITO contact and collected in reflection from the Ag cathode (that is, a double pass in transmission through the active layer) using a Si photodiode. The devices were depleted at −1 V reverse bias and a 300 mVrms, f=1 kHz sinusoidal dither was applied to measure the rms change in transmitted light intensity. The linear absorption data in Fig. 5a of the text were collected in transmission differentially between two films with differing thickness (50 nm test film with a 10-nm reference film) in order to minimize the influence of reflection.
Steady-state EL spectra were collected under direct current bias and PL spectra with 10 mW, λ=532 nm excitation using a fibre-coupled spectrograph with a cooled Si CCD array. Spectrally resolved PL transient data were obtained using a Hamamatsu C5680 streak camera and ~150 fs, 10 μJ cm−2 pump pulses at a pump wavelength of 530 nm and a repetition rate of 2 kHz. The transient spectra provided in Supplementary Fig. 8 confirm that the observed decay is due to emission from the CT state. The intensity of all spectra was calibrated using an Ocean Optics tungsten-halogen calibration lamp and is plotted in units proportional to photon flux/area/time/eV. Appropriate, non-fluorescing longpass filtres were employed in all measurements to avoid interference from grating doubles.
Error bars for the EL and PL emission energies extracted via multipeak fitting reflect uncertainty to ±1 s.d. according to standard nonlinear regression analysis. Error bars associated with dielectric constant, EQE and device performance metrics in the Supplementary Information reflect ±1 s.d. in the result of measurements on multiple different devices with sample sizes ranging from 5 to 10.
Additional information
How to cite this article: Bernardo, B. et al. Delocalization and dielectric screening of charge transfer states in organic photovoltaic cells. Nat. Commun. 5:3245 doi: 10.1038/ncomms4245 (2014).
References
Deibel, C., Strobel, T. & Dyakonov, V. Role of the charge transfer state in organic donor-acceptor solar cells. Adv. Mater. 22, 4097–4111 (2010).
Clarke, T. M. & Durrant, J. R. Charge photogeneration in organic solar cells. Chem. Rev. 110, 6736–6767 (2010).
Bakulin, A. A. et al. The role of driving energy and delocalized states for charge separation in organic semiconductors. Science 335, 1340–1344 (2012).
Jailaubekov, A. E. et al. Hot charge-transfer excitons set the time limit for charge separation at donor/acceptor interfaces in organic photovoltaics. Nat. Mater. 12, 66–73 (2013).
Piliego, C. & Loi, M. A. Charge transfer state in highly efficient polymer-fullerene bulk heterojunction solar cells. J. Mater. Chem. 22, 4141–4150 (2012).
Giebink, N. C., Wiederrecht, G. P., Wasielewski, M. R. & Forrest, S. R. Thermodynamic efficiency limit of excitonic solar cells. Phys. Rev. B 83, 195326 (2011).
Vandewal, K., Tvingstedt, K., Gadisa, A., Inganas, O. & Manca, J. V. Relating the open-circuit voltage to interface molecular properties of donor:acceptor bulk heterojunction solar cells. Phys. Rev. B 81, 125204 (2010).
Rand, B. P., Burk, D. P. & Forrest, S. R. Offset energies at organic semiconductor heterojunctions and their influence on the open-circuit voltage of thin-film solar cells. Phys. Rev. B 75, 115327 (2007).
Giebink, N. C., Wiederrecht, G. P., Wasielewski, M. R. & Forrest, S. R. Ideal diode equation for organic heterojunctions. I. Derivation and application. Phys. Rev. B 82, 155305 (2010).
Loi, M. A. et al. Charge transfer excitons in bulk heterojunctions of a polyfluorene copolymer and a fullerene derivative. Adv. Func. Mater 17, 2111–2116 (2007).
Veldman, D. et al. Compositional and electric field dependence of the dissociation of charge transfer excitons in alternating polyfluorene copolymer/fullerene blends. J. Am. Chem. Soc. 130, 7721–7735 (2008).
Tvingstedt, K., Vandewal, K., Zhang, F. & Inganas, O. On the dissociation efficiency of charge transfer excitons and frenkel excitons in organic solar cells: a luminescence quenching study. J. Phys. Chem. C 114, 21824–21832 (2010).
Piersimoni, F. et al. Influence of fullerene ordering on the energy of the charge-transfer state and open-circuit voltage in polymer: fullerene solar cells. J. Phys. Chem. C 115, 10873–10880 (2011).
Muller, C. et al. Phase behaviour of liquid-crystalline polymer/fullerene organic photovoltaic blends: thermal stability and miscibility. J. Mater. Chem. 21, 10676–10684 (2011).
Madigan, C. F. & Bulovic, V. Solid state solvation in amorphous organic thin films. Phys. Rev. Lett. 91, 247503 (2003).
Sebastian, L., Weiser, G. & Bassler, H. Charge-transfer transitions in solid tetracene and pentacene studied by electro-absorption. Chem. Phys. 61, 125–135 (1981).
Zhang, M., Wang, H., Tian, H., Geng, Y. & Tang, C. W. Bulk heterojunction photovoltaic cells with low donor concentration. Adv. Mater. 23, 4960–4964 (2011).
Kaake, L. G., Sun, Y., Bazan, G. C. & Heeger, A. J. Fullerene concentration dependent bimolecular recombination in organic photovoltaic films. Appl. Phys. Lett. 102, 133302 (2013).
Vandewal, K., Tvingstedt, K., Manca, J. V. & Inganas, O. Charge-transfer states and upper limit of the open-circuit voltage in polymer: fullerene organic solar cells. IEEE J. Sel. Top. Quant. Electron 16, 1676–1684 (2010).
Gould, I. R. et al. Radiative and nonradiative electron-transfer in contact radical-ion pairs. Chem. Phys. 176, 439–456 (1993).
Street, R. A., Davies, D., Khlyabich, P. P., Burkhart, B. & Thompson, B. C. Origin of the tunable open-circuit voltage in ternary blend bulk heterojunction organic solar cells. J. Am. Chem. Soc. 135, 986–989 (2013).
Kohn, P. et al. Crystallization-induced 10-nm structure formation in P3HT/PCBM blends. Macromolecules 46, 4002–4013 (2013).
Scholes, G. D. & Rumbles, G. Excitons in nanoscale systems. Nat. Mater. 5, 683–696 (2006).
Kazaoui, S. et al. Comprehensive analysis of intermolecular charge-transfer excited states in C-60 and C-70 films. Phys. Rev. B 58, 7689–7700 (1998).
Wiederrecht, G. P. et al. Visualizing charge movement near organic heterojunctions with ultrafast time resolution via an induced Stark shift. Appl. Phys. Lett. 100, 113304 (2012).
Pac, B., Petelenz, P., Slawik, M. & Munn, R. W. Theoretical interpretation of the electroabsorption spectrum of fullerene films. J. Chem. Phys. 109, 7932–7939 (1998).
Atkins, P. W. Molecular Quantum Mechanics Oxford University Press (1970).
Lee, J. et al. Charge transfer state versus hot exciton dissociation in polymer-fullerene blended solar cells. J. Am. Chem. Soc. 132, 11878–11880 (2010).
Vandewal, K. et al. Efficient charge generation by relaxed charge-transfer states at organic interfaces. Nat. Mater. 13, 63–68 (2013).
Vithanage, D. A. et al. Visualizing charge separation in bulk heterojunction organic solar cells. Nat. Commun. 4, 2334 (2013).
Jamieson, F. C. et al. Fullerene crystallisation as a key driver of charge separation in polymer/fullerene bulk heterojunction solar cells. Chem. Sci. 3, 485–492 (2012).
Bansal, N. et al. Influence of crystallinity and energetics on charge separation in polymer-inorganic nanocomposite films for solar cells. Sci. Rep. 3, 1531 (2013).
Rao, A. et al. The role of spin in the kinetic control of recombination in organic photovoltaics. Nature 500, 435–439 (2013).
Acknowledgements
This work was supported in part by the DARPA Young Faculty Award and the DOE SunShot program under award number DE-EE0005798. This work was performed, in part, at the Center for Nanoscale Materials, a US Department of Energy, Office of Basic Energy Sciences User Facility under Contract No. DE-AC02-06CH11357.
Author information
Authors and Affiliations
Contributions
B.P.R. fabricated and characterized the devices, D.C. conducted the ellipsometry measurements and B.V. performed the X-ray analysis. R.D.S. measured the photoluminescence transients and B.B. and N.C.G. conducted the electroabsorption, electroluminescence and photoluminescence measurements. All authors contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-8 and Supplementary Reference (PDF 382 kb)
Rights and permissions
About this article
Cite this article
Bernardo, B., Cheyns, D., Verreet, B. et al. Delocalization and dielectric screening of charge transfer states in organic photovoltaic cells. Nat Commun 5, 3245 (2014). https://doi.org/10.1038/ncomms4245
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4245
This article is cited by
-
Selenium substitution for dielectric constant improvement and hole-transfer acceleration in non-fullerene organic solar cells
Nature Communications (2024)
-
Interfacial engineering of Bi2S3/Ti3C2Tx MXene based on work function for rapid photo-excited bacteria-killing
Nature Communications (2021)
-
Influence of static disorder of charge transfer state on voltage loss in organic photovoltaics
Nature Communications (2021)
-
Charge-transfer electronic states in organic solar cells
Nature Reviews Materials (2019)
-
A generalized Stark effect electromodulation model for extracting excitonic properties in organic semiconductors
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
