
Abstract
Sugar beet (Beta vulgaris ssp. vulgaris) is an important crop of temperate climates which provides nearly 30% of the world’s annual sugar production and is a source for bioethanol and animal feed. The species belongs to the order of Caryophylalles, is diploid with 2n = 18 chromosomes, has an estimated genome size of 714–758 megabases1 and shares an ancient genome triplication with other eudicot plants2. Leafy beets have been cultivated since Roman times, but sugar beet is one of the most recently domesticated crops. It arose in the late eighteenth century when lines accumulating sugar in the storage root were selected from crosses made with chard and fodder beet3. Here we present a reference genome sequence for sugar beet as the first non-rosid, non-asterid eudicot genome, advancing comparative genomics and phylogenetic reconstructions. The genome sequence comprises 567 megabases, of which 85% could be assigned to chromosomes. The assembly covers a large proportion of the repetitive sequence content that was estimated4 to be 63%. We predicted 27,421 protein-coding genes supported by transcript data and annotated them on the basis of sequence homology. Phylogenetic analyses provided evidence for the separation of Caryophyllales before the split of asterids and rosids, and revealed lineage-specific gene family expansions and losses. We sequenced spinach (Spinacia oleracea), another Caryophyllales species, and validated features that separate this clade from rosids and asterids. Intraspecific genomic variation was analysed based on the genome sequences of sea beet (Beta vulgaris ssp. maritima; progenitor of all beet crops) and four additional sugar beet accessions. We identified seven million variant positions in the reference genome, and also large regions of low variability, indicating artificial selection. The sugar beet genome sequence enables the identification of genes affecting agronomically relevant traits, supports molecular breeding and maximizes the plant’s potential in energy biotechnology.
Similar content being viewed by others

Main
During the last 200 years of sugar beet breeding, the sugar content has increased from 8% to 18% in today’s cultivars. Breeding has also actively selected for traits like resistance to viral and fungal diseases, improved taproot yield, monogermy of the seed and bolting resistance. After discovering a male sterile cytoplasm, breeders started to develop hybrid varieties and successfully increased yield5. Taxonomy assigns Beta to the Amaranthaceae family within Caryophylalles, an order comprising 11,510 species6 including cacti, ice plants (Aizoaceae), other drought-tolerant species, and carnivorous plants such as pitcher plants (Nepenthes) and sundew (Drosera). Until now, no Caryophyllales species have been sequenced.
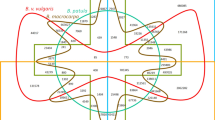
To provide an extended basis for comparative plant genomics and to support molecular breeding, we sequenced the double haploid sugar beet line KWS2320 as reference genotype, using the Roche/454, Illumina and Sanger sequencing platforms (Extended Data Table 1a, Supplementary Table 1). The initial assembly was integrated with genome-wide genetic and physical map information2, resulting in 225 genetically anchored scaffolds (394.6 Mb), assigned to nine chromosomes (Table 1, Fig. 1, Extended Data Figs 1 and 2). The chromosomal nomenclature follows a previous study7 describing a Beta karyotype at chromosome arm resolution. The genetically integrated assembly, ‘RefBeet’, comprised in total 569.0 Mb in 43,721 sequences (2,333 scaffolds and 41,388 unscaffolded contigs) and had an N50 size of 1.7 Mb with 77 scaffolds being of this size or larger. We incorporated Illumina sequencing reads generated from PCR-free libraries and analysed genotyping-by-sequencing data, leading to an optimized assembly of 566.6 Mb in 2,171 scaffolds and 38,337 unscaffolded contigs. The N50 size was 2.01 Mb (the 72nd scaffold) and the chromosomally assigned fraction 84.7% (Table 1). The assembled part of the genome is assumed to represent the unique regions as well as repetitive regions, which are either short enough to be placed in a unique sequence context or divergent enough to behave as unique entities. A k-mer analysis of Illumina data indicated a genome size of 731 Mb (Extended Data Fig. 3a). We located 94% of publicly available isogenic expressed sequence tags (ESTs) in RefBeet, suggesting that gene-containing regions are comprehensively covered. A sequenced bacterial artificial chromosome (BAC) clone8 was compared to the corresponding region in RefBeet and found to be correctly assembled within one scaffold. On average, one mismatch and one insertion or deletion (indel) error occurred in 10 kb. RefBeet resolved regions of recombination suppression in centromeric and pericentromeric regions of chromosomes, flanked by regions showing enhanced recombination rates (Extended Data Fig. 4).

For chromosomes 2–9 see Extended Data Figs 1 and 2. a, Positions of genetic markers in the genetic map2 and the RefBeet assembly. b, Distribution of coding sequence (CDS) and repetitive sequence of the Gypsy type (LTR retrotransposons), the SINE type (non-LTR retrotransposons), the En/Spm type (DNA transposons), and three classes of satellite DNA (intercalary, centromeric, subtelomeric). c, Distribution of mapped small RNAs of 21 and 24 nucleotides (nt). The large peak of 21 nt reads (about 327,000 reads mapped) corresponds to the highly expressed microRNA MIR166. d, Distribution of genomic variants in four sugar beet accessions and sea beet (DeKBm) compared to RefBeet. Shared and individual low-variation regions per accession are visible (for example, region 30–31 Mb is shared among the sugar beet accessions KDHBv, UMSBv, YTiBv, YMoBv).
We identified 252 Mb (42.3%) of RefBeet as repetitive sequence (Supplementary Data 1). The largest group was long terminal repeat (LTR) retrotransposons (Extended Data Fig. 5a). Gypsy-like elements were enriched in centromeric and pericentromeric regions (Fig. 1, Extended Data Figs 1 and 2). Non-LTR retrotransposons of the long interspersed nuclear element (LINE) type were dispersed, whereas short interspersed nuclear elements (SINEs) were enriched towards chromosome ends. Three major satellite classes were organized in large arrays (Fig. 2). By analysing unassembled genomic data we estimated total amounts of 15.4 Mb centromeric, 6.0 Mb intercalary, and 0.6 Mb subtelomeric satellite DNA, as well as 10.0 Mb of 18S-5.8S-25S and 5S ribosomal genes.

a, Chromosomes were stained with 4′,6-diamidino-2-phenylindole (DAPI, blue); large blocks of heterochromatin are visible (arrows). b, In situ hybridization using the major satellites pBV (centromeric, red), pEV (intercalary, green) and pAV (subtelomeric, orange). c, Overlayed images of a and b show the coverage of chromosomes by satellite DNA. Scale bar, 10 μm.
A total of 27,421 protein-coding genes supported by mRNA evidence (Supplementary Table 2) were predicted in RefBeet; 91% included start and stop codons (Supplementary Table 3). The majority of the genes (73.6%) were found within chromosomally assigned scaffolds with on average 5.2 genes per 100 kb, a gene length of 5,252 bp including introns, a coding sequence length of 1,159 bp and 4.9 coding exons per gene. The codon usage was similar to other dicot species (Supplementary Table 4). Homology-based annotation of non-coding RNA genes resulted in 3,005 predictions of tRNAs, microRNAs, small nucleolar RNAs, spliceosomal RNAs and ribosomal RNAs, mainly supported by evidence from isogenic small RNA data (Extended Data Fig. 5b–e, Extended Data Table 1b, Supplementary Table 5).
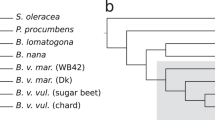
Based on the translated Beta vulgaris gene set and the protein sets of nine other plants (Extended Data Table 1d) we determined 19,747 phylogenetic trees, collectively called ‘phylome’9, and inferred orthologous and paralogous gene relationships (Extended Data Fig. 6a). Previous studies left the phylogeny of rosids, asterids and Caryophyllales unresolved10 or classified Caryophyllales as a subclade of asterids11. A species tree inferred from the collection of gene trees strongly suggested that Beta vulgaris branched off before the separation of asterids and rosids (Fig. 3). Thus, according to our data, Caryophyllales represent the most basal eudicot clade among the studied species. The fraction of species-specific genes within eudicots (Fig. 3) was the largest for sugar beet, reflecting its phylogenetic position. The analysis of paralogous genes provided evidence for the absence of a lineage-specific whole genome duplication in Beta vulgaris supporting previous studies2 (Extended Data Fig. 6b–e, Supplementary Table 6).

Species tree based on maximum-likelihood analysis of a concatenated alignment of 110 widespread single-copy protein sequences (left). The upper bar per species (right with scale on the top) indicates the number of widespread genes that are found in at least 9 of the 10 species (green); eudicot-specific genes that are found in at least 7 of the 8 eudicot species (yellow); species-specific genes with no homologues in other species of this tree (light grey); and remaining genes (brown). The slim bars per species (scale on the bottom) represent the percentage of genes with at least one paralogue (blue) and the percentage of sugar beet genes that have homologues in a given species (pink), respectively.
We functionally annotated 17,151 RefBeet genes (63%) based on sequence homology (Supplementary Data 2). The number of disease resistance genes detected was comparatively small, particularly for the STK-domain containing classes (Supplementary Table 7, Supplementary Data 3). In contrast to previous studies12,13, we found a TNL class resistance gene in the genomes of sugar beet (Bv_22240_ksro) and spinach, both belonging to Amaranthaceae. The phylome tree of Bv_22240_ksro indicated that the presence of a single TNL class gene is a feature of Amaranthaceae, whereas expansion of this gene family is typical for rosids and asterids. The functional categories of expanded and potentially lost gene families (Extended Data Fig. 5f, Supplementary Tables 8, 9) indicate that genes involved in defence and stress compensation represent vital evolutionary targets. The number of transcription factors identified in RefBeet was the lowest of all species studied (Supplementary Table 10). The reference genome sequence enables future experimental approaches to determine if lower gene numbers may alter transcriptional network topologies; Caryophyllales may harbour unknown genes involved in transcriptional control. We identified four sucrose transporter (SUT) orthologues in RefBeet. Phylogenetic analysis including known sucrose transporters suggested a duplication of the SUT1 gene in Amaranthaceae followed by extensive mutation of one paralogue (Extended Data Fig. 7a). The genome sequences of sugar beet and spinach, both containing the four SUT genes, are an excellent basis for studying the implications of this duplication event.
Previous studies addressing the variation within the genus Beta indicated high divergence between genotypes2,14. We generated genome sequences of four non-reference sugar beet double haploid accessions (KDHBv, UMSBv, YMoBv, YTiBv) and characterized the genome-wide variation (Extended Data Table 1a, c, Supplementary Tables 3, 11–13). Within RefBeet we identified 7.0 million positions which were variant (77% substituted, 23% deleted) in at least one of the other accessions and 274.9 million positions which were unchanged in all five accessions. We found 2.9 million variants on average per non-reference accession. Coding regions had a prevalence of indels of length three or multiples of three (44%), compared to non-coding regions (16%). The distribution of variants revealed large regions of low variation (Fig. 1, Extended Data Figs 1, 2, 8a–c). Such variation ‘deserts’ were found in all chromosomes and in all accessions, which might reflect extensive cross-breeding with a limited number of haplotypes in the breeding material, a founder effect, or a bottleneck at the establishment of the crop. However, most of the variation deserts were accession-specific (Extended Data Fig. 8d), probably owing to recombination events that have occurred since the introduction of founder haplotypes into breeding lines. The four accessions shared 50.6 Mb of variation deserts along RefBeet containing 1,824 predicted RefBeet genes (Gene Ontology (GO) term enrichment see Supplementary Table 14). Genes in these regions, analysed in 24 additional sugar beet accessions, showed higher sequence conservation (Extended Data Fig. 8e). These findings suggest that regions of low variation are not maintained by chance, but are rather the result of breeders’ selection towards certain genes contained in those regions. The sea beet Beta maritima is fully interbreedable with sugar beet and commonly used as a valuable source of resistances against biotic or abiotic stress15. We sequenced its genome and identified a total of 75 Mb as variation desert, of which 67 Mb were shared with at least one of the four non-reference Beta vulgaris accessions. These regions may represent traces of breeding activities which aimed at introducing sea beet traits into sugar beet. The gene BvBTC1, encoded by the B-locus16 and located in a 1.1 Mb RefBeet scaffold on chromosome 2, plays an important role during vernalization. Cultivated lines are homozygotes for the B allele resulting in a biennial life cycle. The B-locus is located in variation deserts of all five sugar beet lines, whereas the genome of the annual wild form Beta maritima shows high variation at this locus, demonstrating that breeding has shaped the genome of sugar beet.
Sugar beet is a hybrid crop based on seed pool lines (male steriles, monogerms) and pollen pool lines (pollinators, multigerms). We identified regions of potentially fixed differences between the two groups: the intersection of shared low-variation regions in seed pool lines and shared high-variation regions of identical variation patterns in pollen pool lines comprised 311 genomic regions (1.6 Mb in total) containing 119 genes.
We performed evidence-based gene predictions in the assemblies of KDHBv, UMSBv, YMoBv and YTiBv. Based on the comparison of 2,112 single copy genes, UMSBv had the largest genetic distance to RefBeet (Extended Data Fig. 7b). The number of accession-specific genes ranged from 79 (RefBeet) to 271 (UMSBv). Genes were analysed for the ratio of non-synonymous to synonymous substitutions, altered start and stop sites, new stop codons, modified splice donor or splice acceptor sites and indels, revealing extensive variation in coding regions (Supplementary Tables 15, 16 and Extended Data Fig. 8f). In addition to allelic variation, the variation in gene content may contribute to heterosis, as has been suggested for maize17.
The availability of the sugar beet genome sequence very much simplifies fine-mapping of quantitative trait loci and the discovery of causal genes, as single-nucleotide polymorphism (SNP)-based markers can be designed for any region of the genome. Association mapping to identify regions of shared ancestry in sugar beet lines requires at least 100,000 variant positions for genotyping. Such positions can now be selected from a catalogue of seven million variants. The genome sequence facilitates further experimentation to characterize gene functions, which accelerates the identification of rewarding targets for transgenic manipulation, and represents an important foundation for molecular and comparative studies in sugar beet, Caryophyllales and flowering plants. The data presented are key to improvements of the sugar beet crop with respect to yield and quality and towards its application as a sustainable energy crop.
Methods Summary
Genome sequencing and assembly
Genomic DNA isolated from root and leaf material was sequenced on the Roche/454 FLX, Illumina HiSeq2000 and ABI3730 XL sequencing platforms. The Newbler software was applied on 454, Illumina and Sanger sequencing data to assemble the reference genotype (RefBeet). Contigs of putative bacterial origin and those smaller than 500 bp were removed. Additional lines were sequenced on the HiSeq2000 platform and were assembled using SOAPdenovo. We performed gap-closing and homopolymer error correction using Illumina reads from PCR-based and PCR-free libraries (Extended Data Fig. 3b, c). Chromosome-wise scaffolding using genetic and physical mapping data was assisted by SSPACE (Methods and Supplementary Methods).
Gene annotation
Prediction of protein coding genes was performed using the AUGUSTUS pipeline, with Illumina mRNA-seq reads and other cDNA read data as supporting evidence. Gene models were filtered for transposable element homology. Small and other non-coding RNAs were identified based on homology searches and based on Illumina sequencing data. Repeats were predicted using RepeatModeler, followed by manual curation of the predictions (Methods and Supplementary Methods).
Intraspecific variation
Variant positions (substitutions, indels) were identified by read mapping and scaffold alignment (Methods and Supplementary Methods).
Phylogenetic analysis and species tree reconstruction
The longest protein sequence of each RefBeet gene was used for a Smith–Waterman search against the protein sets of nine other plant species. Alignments were generated and quality-filtered, and phylogenetic trees were calculated for each Beta vulgaris sequence. A species tree was generated from a super-tree of all trees and by multi-gene phylogenetic analysis of high-confidence 1:1 orthologues.
Functional annotation
Protein coding gene predictions were functionally annotated based on protein signatures and orthology relationships.
Online Methods
Sequencing and assembly
Genomic DNA isolated from root and leaf material was sequenced on the Roche/454 FLX, Illumina HiSeq2000 and ABI3730 XL sequencing platforms. The plant material included five double haploid and two inbred sugar beet breeding lines (Beta vulgaris ssp. vulgaris; referred to as Beta vulgaris), one wild beet accession (Beta vulgaris ssp. maritima; referred to as Beta maritima), and one spinach accession (Spinacia oleracea). Additionally, 15 genotypes from an F2 panel of a Beta vulgaris cross used to generate beet genetic maps2,14 were sequenced at low coverage (Supplementary Methods).
Illumina genomic sequencing was performed on a HiSeq2000 sequencing instrument with 2 × 100 nt for paired-end reads and 2 × 50 nt for mate-pair reads (Supplementary Tables 1 and 11). Roche/454 genomic single-read and mate-pair sequencing was performed on a Roche/FLX sequencing instrument using Titanium XLR70 sequencing kits (Roche/454 Life Sciences). End-sequencing of genomic BAC and fosmid libraries introduced previously2,18,19 was performed on an ABI3730 XL DNA Analyzer. Genomic Roche/454 and Illumina data, BAC ends and fosmid ends were filtered for low-quality sequence, contamination and redundancy, and all data sets were assembled together on a 512 GB access-random memory (RAM) computer using the Newbler software (v2.6 20110630_1301, parameters -fe exclude_list -siod -nrm -scaffold -large -ace -ar -a 40 -l 500 -cpu 48).
We removed potential bacterial contigs and scaffolds based on three criteria: a GC content of 60% or higher, the presence of predicted genes without sugar beet cDNA support, and the absence of both sugar beet repeats and genes supported by cDNA data. The assembly size was determined by adding up the lengths of the scaffolds and the lengths of additional unscaffolded contigs larger than 500 bp (smaller contigs were removed). The N50 size refers to this assembly size as 100% and reports the length of the scaffold or contig that spans the 50% mark after sorting the sequences by length.
Illumina-only assemblies were performed on 100 GB and 256 GB RAM computers using the SOAPdenovo20 software v1.05 (SOAPdenovo-63mer, -K 49, pair_num_cutoff = 3, map_len = 32) followed by gap filling using GapCloser v1.12.
mRNA-seq sequencing was carried out on the Illumina Genome Analyzer (GA) IIx and Illumina HiSeq2000 sequencing instruments. From each library, one lane of data was generated with read lengths of 2 × 54 nt on the GA and 2 × 50 nt on the HiSeq2000. Small RNA libraries were sequenced with 36 nt reads on the GA and 50 nt reads on the HiSeq2000. Additional cDNA sequences from sugar beet were generated by Roche/454 Life Sciences on the GS20 platform with an average length of 106 bp. The transcript data generated are summarized in Supplementary Table 2.
Integration with genetic and physical maps as well as genotyping-by-sequencing (GBS) data
Sequence information of anchored markers in genetic and physical maps2 was used to assign scaffolds to chromosomes and to build connections between scaffolds. Confirmation and further genetic integration was derived from generating and analysing GBS data (see Supplementary Methods). To establish new connections between scaffolds, a group of scaffolds placed as neighbours based on genetic integration was used as input for SSPACE21 (v1.1, parameters -x 0 -k 1) together with the six largest paired data sets (Illumina 6 kb, Roche/454 7 kb, 10 kb and 20 kb, fosmid ends, BAC ends). The output was manually corrected if necessary. Analyses and control steps were coded in Perl v5.8.9 or used UNIX shell commands.
Correction of small indels in the assembly and gap closing
For consensus sequence correction we mapped quality filtered Illumina reads (2 × 100 nt, 93-fold genome coverage) against the assembly using BWA22 v0.5.9 allowing for 3 edits. Indels were identified using SAMtools mpileup23 v0.1.18. A total of 9,101 bp insertions and 60,685 bp deletions were corrected in the consensus sequence. Indel errors were corrected if error positions were covered by at least 10 reads, if at least 60% of them showed the same indel and if the indel was confirmed on both strands. The criteria were validated using the Sanger sequence of the sugar beet BAC insert ZR47B15 (ref. 8).
Gap closing was performed using 670 million Illumina paired-end reads generated from two PCR-amplified libraries with insert sizes 600 nt and 250 nt, and from five PCR-free libraries with insert sizes 200–700 nt as input for GapCloser20 (v1.12-r6, default parameters and -p set to 31).
Annotation of repetitive elements
The de novo identification and classification of repeats within the RefBeet assembly was performed using RepeatModeler (http://www.repeatmasker.org). RepeatModeler v1.0.5 was installed along with RECON24 v1.07, RepeatMasker ‘open 3-3-0’, and RMBlast v1.2 with BLAST v2.2.23 (all loaded from http://www.repeatmasker.org), RepeatScout25 v1.0.5 (http://bix.ucsd.edu/repeatscout/), Tandem Repeat Finder26 (trf404 loaded from http://tandem.bu.edu/), and the RepeatMasker libraries (http://www.girinst.org/server/RepBase/) as of September 2011. RepeatModeler was applied on the database file created by the BuildDatabase subprogram which was run on the RefBeet assembly. The output was used as library for RepeatMasker (parameters -e crossmatch -pa 20 -gff) to generate a masked version of the assembly and to get the genomic positions of the repeat annotation. The repetitive fraction of the assembly was determined based on the RepeatMasker output. The automated repeat classification provided by RepeatModeler was refined by manual curation of the data (see Supplementary Methods). The repeat families along with the combined automated and manual classification are listed in Supplementary Data 1. The fractions of different repeat classes annotated in RefBeet are shown in Extended Data Fig. 5a.
The distribution of small RNAs (Fig. 1 and Extended Data Figs 1 and 2) was analysed by mapping 677.8 million adaptor-trimmed small RNA sequences of three libraries (see Supplementary Table 5) against RefBeet using BWA v0.6.1. Reads shorter than 15 bases after trimming were removed. The mapping seed length was set to 15. If a read mapped to multiple locations one random location was kept. Custom Perl scripts were used to locate mapped reads within the annotation of non-coding RNAs. The read length distribution and the chromosome-wide distribution of mapped reads were computed with Perl and plotted with R v2.15.
Coding gene prediction
The evidence-based de novo annotation of coding genes was performed applying the program AUGUSTUS27 v2.5.5 on the RefBeet assembly. The evidence was provided by 616.3 million filtered Illumina mRNA-seq reads (mainly generated as paired-ends) from five Beta vulgaris accessions and different tissues, 282,169 cDNA single-end reads generated on a Roche/454 GS20 platform, and 35,523 EST sequences from public databases. The Roche/454 reads and most of the ESTs were derived from genotype KWS2320. AUGUSTUS settings were: using Arabidopsis training data, reporting untranslated regions in addition to the coding sequences, reporting alternative transcripts if suggested by hints, and accepting introns that start with AT and end with AC in addition to introns with starts flanked by GT-AG and GC-AG.
For each Beta vulgaris accession we initially predicted 30,339 to 36,589 genes. After removal of (retro)transposon gene candidates, the gene sets used for downstream analyses consisted of 25,368 to 31,355 genes (Supplementary Table 3). Of those genes, 77–94% had both start and stop codon, and the fraction of predictions completely supported by cDNA was 48–61%.
We identified transposable element-related genes in the automated gene prediction of RefBeet by screening the phylomes for GO terms specific to transposable elements, by running the program ‘TransposonPSI’ (http://transposonpsi.sourceforge.net/), and by analysing the genomic positions of the repeat annotation (Supplementary Methods). In total, 4,643 transposable element candidates were removed from the initial set of 32,064 evidence-based genes predicted in RefBeet by AUGUSTUS.
The predicted genes of assemblies generated with SOAPdenovo (Supplementary Tables 3, 11) were screened for overlap with sequences of transposable elements contained in the repeat annotation. The assemblies were masked using RepeatMasker, and gene predictions were omitted from further analyses if at least one base of their coding parts overlapped with an annotated transposable element.
Plant species analysed in comparative studies
Comparative analyses were carried out based on data from seven dicot (five rosids and two asterids) and two monocot species: Arabidopsis thaliana, Glycine max, Populus trichocarpa, Theobroma cacao, Vitis vinifera, Solanum lycopersicum, Solanum tuberosum, Oryza sativa ssp. indica and Zea mays (Extended Data Table 1d, Supplementary Methods).
Annotation of non-coding RNA genes
Non-coding RNA genes were predicted using the programs tRNAscan-SE28, RNammer29, and BLAST30, and based on database searches in Rfam31, the plant snoRNA database32, GenBank33, and the ASRG database34. To support the predictions, we mapped 677.8 million Illumina small RNA reads generated from root, inflorescence and leaf material of the reference genotype against RefBeet (Supplementary Table 5). Reads were adaptor trimmed (custom Perl script) and mapped using BWA v0.6.1 with a seed length of 15 bases and one edit allowed in the alignment. The cDNA coverage was determined with SAMtools mpileup and custom Perl scripts.
Phylome reconstruction and orthology/paralogy predictions
The longest protein sequence for each gene annotated in RefBeet was used for a Smith-Waterman search (E-value cutoff 1 × 10−5, matching length >50% of the query sequence) against the protein sets of nine other species. Alignments were generated and quality-filtered, and phylogenetic trees were calculated for each Beta vulgaris sequence (see Supplementary Methods). The collection of phylogenetic trees is referred to as the sugar beet ‘phylome’, based on which we inferred the orthologous and paralogous relationships of the genes by considering each node as either a speciation or duplication event.
Species tree reconstruction
A phylogeny describing the evolutionary relationships of the species included in the phylome was inferred using two complementary approaches resulting in identical tree topologies (Fig. 3). First, a super-tree was inferred from all the trees in the phylome (19,747 trees) by using a gene tree parsimony approach as implemented in the DupTree algorithm35. This approach is different from other super-tree approaches (such as finding the majority-rule consensus) as it finds the species topology with the minimum total number of duplications implied when reconciling a collection of gene family trees (that is, the phylome) with that species topology. Second, 110 gene families with high-confidence one-to-one orthology in at least 9 of the 10 species were used to perform a multi-gene phylogenetic analysis. Protein sequence alignments were performed as described (see phylome reconstruction in Supplementary Methods) and concatenated into a single alignment. Species relationships were inferred from this alignment using a maximum likelihood (ML) approach as implemented in PhyML36 using the Jones–Taylor–Thornton (JTT) evolutionary model; for 97 of 110 gene families this model was best-fitting. Branch supports were computed using an aLRT (approximate likelihood ratio test) parametric test based on a chi-square distribution. Both complementary approaches resulted in an identical topology. Such congruence is suggestive that a correct phylogeny was found.
To track specific or shared genes in the species tree an all-against-all BLAST search of the protein sets of the ten species was performed (E-value cutoff 1 × 10−5). The patterns of homology across species and clades were computed. The result was categorized as widespread genes, eudicot-specific genes and species-specific genes (Fig. 3).
Whole genome duplication analysis using collinear blocks of coding genes
We performed a Ks analysis of 370 paralogous gene pairs forming 34 collinear blocks (Extended Data Fig. 6c–e). Collinear blocks of coding genes were determined using MCScanX37 applied on the RefBeet protein set (longest protein isoform per gene). Protein sequences were aligned against themselves using BLASTp, the top 5 alignments per gene were kept. High-confidence collinear blocks with an E-value lower than 1 × 10−10and a score larger than 300 were selected (parameters suggested by MCScanX). A total of 34 blocks of 7–35 gene pairs (on average 11 pairs, in total 370 pairs) were found. Ks values were calculated using MCScanX, which implements the Nei-Gojobori algorithm38
Functional annotation of protein coding genes
Protein coding gene predictions were functionally annotated based on protein signatures and orthology relationships. In the protein signature approach, each sugar beet protein was inspected for different signatures such as families, regions, domains, repeats, and binding sites using InterProScan39 v4.8 and a set of different databases (PROSITE, PRINTS, Pfam, ProDom, SMART, TIGRFAMs, PIR superfamily, SUPERFAMILY, Gene3D, PANTHER, HAMAP). Additionally, BLAST searches against SwissProt40, KEGG41, and KOG42 databases were performed, and annotations were extracted. In the phylogeny-based approach 15,263 one-to-one orthology relationships between Beta vulgaris genes and GO-annotated genes of other plant species were inferred from trees in the sugar beet phylome. The annotations from all sources were combined, and a merged annotation table was generated (Supplementary Data 2).
We predicted and classified resistance gene analogue (RGA) genes by applying a modified version of an HMM-based pipeline43 (Supplementary Methods). The numbers of RGAs detected in Beta vulgaris and nine other plant species are listed in Supplementary Table 7. The gene identifiers of all putative RGAs in the Beta vulgaris genome are listed in Supplementary Data 3. Proteins were classified into 56 transcription factor families and subfamilies based on protein domains defined by InterPro motives44 indicating DNA binding or other domains characteristic to transcription factors45 (see Supplementary Methods). The numbers of transcription factors identified per species and per transcription factor class are listed in Supplementary Table 10. Sucrose transporter (SUT) proteins were identified by comparison of 40 known SUT protein sequences of 15 higher plants46 against RefBeet (see Supplementary Methods).
Expanded and potentially lost gene families in Beta vulgaris
Expanded gene families were detected by searching the Beta vulgaris phylome for genes specifically duplicated in Beta vulgaris. To determine potentially lost gene families in Beta vulgaris the set of protein sequences of Arabidopsis was used for a BLAST search (E-value cutoff 1 × 10−5, minimum 50% length of the query protein) against Beta vulgaris proteins.
Intraspecific variation
Four non-reference sugar beet accessions were sequenced (KDHBv, UMSBv, YMoBv, YTiBv). Additional data for the reference accession, processed in the same way, was generated as a control and quality measure (referred to as ‘RefBv’). We generated a merged variant collection from RefBeet positions covered by read mapping or scaffold alignment and distinguished positions that were identical, variant (substituted or deleted), or uncalled (‘N’ in either RefBeet or in the other accession’s assembly). The number of insertions, deletions, substitutions and mixed events (indel plus substitution) was counted.
Substitutions, insertions and deletions contained in coding regions were extracted, counted and categorized using a custom Perl script. Categories were indels, synonymous and non-synonymous codon alterations, changed start and stop codons, new stop codons, and splice donor or acceptor sites alterations (Supplementary Table 15). Splice sites were considered as altered if variants affected the first two or the last two bases of an intron. The standard genetic code was used to translate the coding sequence into amino acids and stop codons. The number of transitions (A G, T
G, T C), transversions (A
C), transversions (A T, C
T, C G, A
G, A C, G
C, G T), and indels of length three or multiples of three (Extended Data Fig. 8f) were determined using a custom Perl script.
T), and indels of length three or multiples of three (Extended Data Fig. 8f) were determined using a custom Perl script.
We discovered regions in the genome with low variant rates (≤2 variants per 2 kb window) which we refer to as variation deserts (Extended Data Fig. 8a–d). Variant positions, identical positions and excluded positions (mainly due to low coverage) were counted per 2 kb intervals (shifted by 1 kb). An interval was considered as desert interval if at most two variants and at most 500 excluded bases were contained. A variant desert was defined as stretch of adjacent desert intervals. Genes located within a variant desert with at least 90% of the genomic length (CDS and UTRs) were considered as variation desert genes. We determined the frequency of each GO term assigned to the group of 1,824 desert genes and the remaining 25,597 genes. GO terms were kept if they were at least ten times more frequent within the group of desert genes (that is, GO terms with odds ratio <10 were removed). The probability that the enrichment occurred by chance was calculated using the two-tailed Fisher exact test (P value cutoff 0.05, no correction for multiple testing). Enriched GO terms are listed in Supplementary Table 14. The conservation of 51 RefBeet genes inside and outside of shared variation deserts was measured by screening for polymorphisms within an extended panel of 24 sugar beet genotypes representing different breeding programs (Supplementary Methods).
To analyse the presence or absence of RefBeet genes within the genomes of other double haploid accessions, Illumina paired-end reads of KDHBv, UMSBv, YTiBv, and YMoBv were mapped against the RefBeet assembly using BWA v0.5.9 (3 edits allowed). Only uniquely mapping reads were considered. Before inferring absence or presence of a gene in the non-reference accessions, the coverage of RefBeet genes was confirmed by mapping Illumina data of the reference genotype: a CDS part of RefBeet genes was ignored if less than 90% of its length was covered by RefBv reads (10.6% of 27,421 RefBeet genes entirely ignored). Genes were considered absent in one of the other accessions if less than 1% within the total of retained CDS length was matched by reads from the non-reference accession. To detect accession-specific genes, the procedure was performed for each accession separately.
The phylogenetic tree of the sugar beet accessions was constructed based on a set of 2,112 single copy genes shared between the five sugar beet accessions. The protein sequences were used to generate multiple alignments based on which a phylogenetic tree was constructed (Extended Data Fig. 7b).
Accession codes
Accessions
BioProject
GenBank/EMBL/DDBJ
Sequence Read Archive
Data deposits
Sequencing raw data (genomic and transcript sequences) have been submitted to the SRA archive with the study accession number SRP023136. The NCBI Bioproject accession is PRJNA41497. The whole-genome shotgun assemblies have been deposited at DDBJ/EMBL/GenBank under the accessions AYZS00000000–AYZY00000000. The GenBank accession numbers KG026656–KG039419 were assigned to BAC end sequences and JY274675 –JY473858 to fosmid end sequences generated in this study. Plant material for Beta vulgaris genotype KWS2320 and Beta maritima 9W_2101 (DeKBm) are available as seeds by signing a material transfer agreement (MTA). A sugar beet website including a genome browser has been set up at http://bvseq.molgen.mpg.de, providing access to assemblies, annotations, gene models and variation data. The sugar beet phylome can be accessed at http://phylomeDB.org.
References
Arumuganathan, K. & Earle, E. D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218 (1991)
Dohm, J. C. et al. Palaeohexaploid ancestry for Caryophyllales inferred from extensive gene-based physical and genetic mapping of the sugar beet genome (Beta vulgaris). Plant J. 70, 528–540 (2012)
Fischer, H. E. Origin of the ‘Weisse Schlesische Rübe’ (white Silesian beet) and resynthesis of sugar beet. Euphytica 41, 75–80 (1989)
Flavell, R. B., Bennett, M. D., Smith, J. B. & Smith, D. B. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet. 12, 257–269 (1974)
Biancardi, E., McGrath, J. M., Panella, L., Lewellen, R. & Stevanato, P. in Root Tuber Crops Vol. 7 (ed. Bradshaw, J. E. ) 173–219 (Springer, 2010)
Stevens, P. Angiosperm Phylogeny Website (2012) http://www.mobot.org/MOBOT/research/APweb/
Paesold, S., Borchardt, D., Schmidt, T. & Dechyeva, D. A sugar beet (Beta vulgaris L.) reference FISH karyotype for chromosome and chromosome-arm identification, integration of genetic linkage groups and analysis of major repeat family distribution. Plant J. 72, 600–611 (2012)
Dohm, J. C., Lange, C., Reinhardt, R. & Himmelbauer, H. Haplotype divergence in Beta vulgaris and microsynteny with sequenced plant genomes. Plant J. 57, 14–26 (2009)
Huerta-Cepas, J., Dopazo, H., Dopazo, J. & Gabaldón, T. The human phylome. Genome Biol. 8, R109 (2007)
The Angiosperm Phylogeny Group An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot. J. Linn. Soc. 141, 399–436 (2003)
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G. & Soltis, D. E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl Acad. Sci. USA (2010)
Hunger, S. et al. Isolation and linkage analysis of expressed disease-resistance gene analogues of sugar beet (Beta vulgaris L.). Genome 46, 70–82 (2003)
Tian, Y., Fan, L., Thurau, T., Jung, C. & Cai, D. The absence of TIR-type resistance gene analogues in the sugar beet (Beta vulgaris L.) genome. J. Mol. Evol. 58, 40–53 (2004)
Schneider, K. et al. Analysis of DNA polymorphisms in sugar beet (Beta vulgaris L.) and development of an SNP-based map of expressed genes. Theor. Appl. Genet. 115, 601–615 (2007)
Biancardi, E., Panella, L. W. & Lewellen, R. T. Beta maritima: The Origin of Beets (Springer, 2012)
Pin, P. A. et al. The role of a pseudo-response regulator gene in life cycle adaptation and domestication of beet. Curr. Biol. 22, 1095–1101 (2012)
Schnable, P. S. & Springer, N. M. Progress toward understanding heterosis in crop plants. Annu. Rev. Plant Biol. 64, 71–88 (2013)
Hohmann, U. et al. A bacterial artificial chromosome (BAC) library of sugar beet and a physical map of the region encompassing the bolting gene B. Mol. Genet. Genomics 269, 126–136 (2003)
Lange, C., Holtgräwe, D., Schulz, B., Weisshaar, B. & Himmelbauer, H. Construction and characterization of a sugar beet (Beta vulgaris) fosmid library. Genome 51, 948–951 (2008)
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012)
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009)
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002)
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005)
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999)
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008)
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997)
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007)
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990)
Burge, S. W. et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 41, D226–D232 (2013)
Brown, J. W. S. et al. Plant snoRNA database. Nucleic Acids Res. 31, 432–435 (2003)
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Wheeler, D. L. GenBank: update. Nucleic Acids Res. 32, D23–D26 (2004)
Wang, B.-B. & Brendel, V. The ASRG database: identification and survey of Arabidopsis thaliana genes involved in pre-mRNA splicing. Genome Biol. 5, R102 (2004)
Wehe, A., Bansal, M. S., Burleigh, J. G. & Eulenstein, O. DupTree: a program for large-scale phylogenetic analyses using gene tree parsimony. Bioinformatics 24, 1540–1541 (2008)
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010)
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012)
Nei, M. & Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426 (1986)
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001)
UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75 (2012)
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012)
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003)
Kim, J. et al. A genome-wide comparison of NB-LRR type of resistance gene analogs (RGA) in the plant kingdom. Mol. Cells 33, 385–392 (2012)
Hunter, S. et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40, D306–D312 (2012)
Young, N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011)
Kühn, C. & Grof, C. P. L. Sucrose transporters of higher plants. Curr. Opin. Plant Biol. 13, 287–297 (2010)
Acknowledgements
This work was supported by the BMBF grant “Verbundprojekt GABI BeetSeq: Erstellung einer Referenzsequenz für das Genom der Zuckerrübe (Beta vulgaris)”, FKZ 0315069A and 0315069B (to H.H. and B.W.) and by the BMBF grant “AnnoBeet: Annotation des Genoms der Zuckerrübe unter Berücksichtigung von Genfunktionen und struktureller Variabilität für Nutzung von Genomdaten in der Pflanzenbiotechnologie.”, FKZ 0315962 A, 0315962 B and 0315962 C (to B.W., H.H., and T.S.). We are grateful to M. Zehnsdorf, H. Kang, P. Viehoever, E. Castillo, A. Menoyo and C. Lange for library preparation and sequencing; to D. Datta for sequencing data base calling; to D. Kedra for discussions; and to D. Boyd and M. Isalan for language editing. We thank P. Pin, B. Briggs, and Strube Research for providing plant material and for discussions. We thank Roche for data generation on the 454 sequencing platform (cDNA and genomic 20 kb mate-pairs) and for early access to the Newbler genome assembly software.
Author information
Authors and Affiliations
Contributions
H.H., B.W. and J.C.D. conceived the study, H.H., D.H., T.R.S., R.R. and B.W. prepared sequencing data, J.C.D., A.E.M., D.H., S.C.-G., F.Z., H.T., O.R., R.S., A.G., B.S., T.K., P.F.S., T.S. and T.G. designed experiments and analysed the data, H.L. participated in project design, J.C.D., H.H. and A.E.M. wrote the paper with input from all other authors. All authors have read and have approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Genomic features of RefBeet chromosomes 2–5.
Section 1 shows the positions of genetic markes in the genetic map2 and RefBeet. Section 2 shows the distribution (stacked area graphs) of predicted coding sequence (CDS) and repetitive sequence of the Gypsy type (LTR retrotransposon), the SINE type (non-LTR retrotransposon), the En/Spm type (DNA transposon), and three classes of satellite DNA (intercalary, centromeric, subtelomeric). The number of bases per feature is displayed in windows of 500 kb (shifted by 300 kb). Section 3 shows the distribution (stacked area graphs) of mapped small RNAs of length 21 and 24 nt in adjacent bins of 500 kb. For reads mapping at multiple locations, one random location was selected. Reads matching within predicted rRNA loci were ignored. Positions with more than 10 thousand mapped 21 nt sequences were labelled with the corresponding non-coding RNA prediction, if available, including the number of matching reads. Section 4 shows the chromosome-wide distribution of genomic variants in four sugar beet accessions and sea beet compared to RefBeet. Substitutions and deletions were detected by read-mapping with up to three variants per 100 nt read in 50 kb windows shifted by 25 kb. Shared and individual low-variation regions per accession are visible.
Extended Data Figure 2 Genomic features of RefBeet chromosomes 6–9.
For details see Extended Data Fig. 1.
Extended Data Figure 3 K-mer distribution and read coverage.
a, Number of 17mers at different coverages. b, c, Correlation of read coverage and GC content of reads generated from a PCR-amplified library (b) and a PCR-free library (c). Read data sets in b and c were aligned against RefBeet. The GC content and the amount of aligned bases were computed in sliding windows of 500 bases shifted by 100 bases. To reduce the amount of data points only chromosome 1 scaffolds 1 (Bvchr1.sca001, 8 Mb) was plotted.
Extended Data Figure 4 Genetic vs physical distances.
a, Genetic and physical positions of 983 genetic markers in the genetic map of sugar beet2 and the RefBeet assembly, respectively. The expected physical distance in sugar beet had been reported as 855 kb per 1 cM, with deviations of up to 50-fold2. In RefBeet only 5% of marker pairs showed the expected physical distance (855 kb ± 20%) suggesting strict partitioning of the genome into regions favouring or disfavouring recombination events.
Extended Data Figure 5 Annotation of repeats and non-coding RNA genes.
a, Repeat content of the sugar beet genome assembly. A total of 252 Mb (42.3%) of the genome assembly consist of repetitive DNA with retrotransposons as the most abundant repeat fraction. All major superfamilies of DNA transposons were represented, showing a dispersed or slightly centromere-enriched distribution along the chromosomes. Microsatellites and minisatellites were well represented owing to flanking heterogeneous sequences, which allowed their assembly. The remaining repetitive sequences (‘Unknown’) in 459 families represent potentially new repeats, most likely rearranged or truncated retrotransposons. b, Summary of non-coding RNA gene annotations. For different classes (miRNA, microRNA; rRNA, ribosomal RNA; snRNA, spliceosomal RNA; snoRNA, small nucleolar RNA; tRNA, transfer RNA) the number of predictions, the number and percentage of predictions with overlapping small RNA reads, and the number of families/subtypes are listed. c, Proportion of annotated tRNAs by amino acid for the five species studied (At, Arabidopsis thaliana; Pt, Populus trichocarpa; Bv, Beta vulgaris; Vv, Vitis vinifera; Zm, Zea mays). d, Absolute numbers of annotated tRNAs by amino acid. Except for pseudogenes the proportion of tRNAs is relatively constant among all species (species names as in c). e, Number of annotated tRNAs by amino acid and species (as predicted by tRNAscan-SE). The total is computed without the last two rows containing pseudogenes and presumably defunct tRNAs with undetermined anti-codon. f, Number and size of Beta vulgaris gene clusters of at least 10 members representing expanded gene families. A total of 1,274 genes are contained in 97 clusters.
Extended Data Figure 6 Analysis of paralogous and orthologous genes.
a, Number and percentage of detected orthologues between Beta vulgaris and nine other plant species. Orthology relationships with 10 or more proteins for any of the species were discarded in order to avoid biases introduced by species-specific gene family expansions. In total, 18,927 sugar beet genes had orthologues in at least one of nine plants, and 16,062 paralogous sugar beet genes appeared in 14,852 trees. b, Number of duplication events detected in gene trees grouped into three age classes. The duplication ratio was calculated as the number of age class-specific duplication events divided by the total number of trees containing duplication events. c, d, Collinear blocks of protein coding genes in Beta vulgaris as dotplot (c) or circular plot (d). Each dot or connecting line represents one gene pair, respectively. Shown are 34 collinear blocks containing 7–35 gene pairs. A triplicated region is visible on chromosomes 1, 3 and 5 (arrows). e. Histogram of Ks values for Beta vulgaris protein coding gene pairs in collinear blocks. Ks values mainly scatter between 1.2 and 1.8 and show peaks at 1.2 and 1.7.
Extended Data Figure 7 Phylogenetic trees.
a, Phylogenetic tree of 44 sucrose transporter protein sequences in higher plants including Beta vulgaris. The reliability for internal branches is indicated in red ranging from 0 = unreliable to 1 = highly reliable (aLRT statistics). At, Arabidopsis thaliana; Bv, Beta vulgaris; Dc, Daucus carota; Hb, Hevea brasiliensis; Hv, Hordeum vulgare; Le, Lycopersicum esculentum renamed Solanum lycopersicum; Lj, Lotus japonicus; Lp, Lolium perenne; Nt, Nicotiana tabacum; Os, Oryza sativa; Ps, Pisum sativum; Sh, Saccharum Hybrid Cultivar Q117; St, Solanum tuberosum; Sb, Sorghum bicolor; Ta, Triticum aestivum; Zm, Zea mays. b, Intraspecific relationship of five Beta vulgaris accessions based on the alignment of 2,112 shared genes.
Extended Data Figure 8 Intraspecific variation.
a, Number of variants inferred from read mapping (black) and sequence identity of matching scaffolds (blue) along RefBeet chromosome 7. The variation profiles of five different accessions including the reference accession (RefBv) are shown. Regions with a high number of read-mapping variants showed a higher density of scaffolds of low sequence identity. However, low-identity scaffolds were also present in low-variation regions of mapped reads. b, Detailed view of the distribution of read mapping variants and read coverage (green) in Beta vulgaris accession KDHBv compared to RefBeet. The secondary y axis on the right side indicates the percentage of positions per window covered in the alignment. Low-variation regions were generally well covered. c, Fraction of variation deserts of different lengths along RefBeet based on read-mapping of genomic data sets and alignment of assembled scaffolds. The six different genotypes include the reference, four other Beta vulgaris accessions, and one Beta maritima accession. Variation deserts were found in all chromosomes. The variation deserts of non-reference sugar beet accessions contained 49% (179 Mb in KDH) to 58% (217 Mb in YMo) of all covered RefBeet positions. d, Intersection of variant deserts. Starting from RefBv, the size of shared variant deserts decreased by including additional Beta vulgaris accessions. e, Sequence conservation comparison of three groups of genes. Genes with GO term enrichment localized within variation deserts shown in red; genes without GO term enrichment localized within variation deserts shown in blue, genes localized outside of variation deserts shown in green. For each of the three groups 17 randomly selected genes with confirmed exon–intron structure were aligned to 24 additional sugar beet accessions. The sequence conservation was determined from the identity of the sequence alignment. Genes with GO term enrichment localized within variation deserts had the highest fraction of high identity gene alignments, followed by genes without GO term enrichment localized within variation deserts. f, Length distribution of insertions and deletions in coding sequences. Apart from one-base indels, indels of length three or multiples of three (3n) were overrepresented. Of all genes affected by indels, 49.1% had a single 3n indel and 5.0% had more than one indel (any length) with bases summing up to 3n.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Tables 1-16, Supplementary Notes and additional references. (PDF 2035 kb)
Supplementary Data 1
Repeat families as detected by RepeatModeler along with the combined automatic and manual classification. (XLS 110 kb)
Supplementary Data 2
Predicted RefBeet genes and their functional annotation based on database searches and transfer from orthologs. (XLS 8639 kb)
Supplementary Data 3
List of 715 putative resistance gene analogs (RGA). Beta vulgaris (Bv) genes were classified based on the presence of RGA domains (columns A+B). In 30 additional Bv genes (column D) these domains were missing in exon parts, but the genes showed sequence homology with known RGAs from other plants. (XLS 69 kb)
Source data
Rights and permissions
This work is licensed under a Creative Commons Attribution-Non-Commercial-ShareAlike 3.0 Unported licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Dohm, J., Minoche, A., Holtgräwe, D. et al. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 505, 546–549 (2014). https://doi.org/10.1038/nature12817
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12817
This article is cited by
-
High-fidelity (repeat) consensus sequences from short reads using combined read clustering and assembly
BMC Genomics (2024)
-
Genome-wide identification, evolution, and role of SPL gene family in beet (Beta vulgaris L.) under cold stress
BMC Genomics (2024)
-
Genotype by environment and genotype by yield*trait interactions in sugar beet: analyzing yield stability and determining key traits association
Scientific Reports (2024)
-
Genome-Wide Identification of Sugar Beet (Beta vulgaris L.) MAPKKKs Gene Family and Their Expression in Response to Salt Stress
Sugar Tech (2024)
-
The role of tissue succulence in plant salt tolerance: an overview
Plant Growth Regulation (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
