
Abstract
Monomorphic post-transplant lymphoproliferative disorder commonly resembles diffuse large B-cell lymphoma or Burkitt lymphoma, and most are Epstein–Barr virus (EBV) positive. We retrospectively identified 32 cases of monomorphic post-transplant lymphoproliferative disorder from two institutions and evaluated EBV in situ hybridization; TP53 mutation status; p53, CD30, myc, and BCL2 expression by immunohistochemistry; proliferation index by Ki67; and germinal center vs non-germinal center immunophenotype by Hans criteria. Post-transplant lymphoproliferative disorder arose after hematopoietic stem cell transplant in five and solid organ transplant in 27 patients, a median of 4 and 96 months after transplant, respectively (overall median latency 71 months, range 2–295). The most common morphology was diffuse large B-cell lymphoma (28 cases), with three cases of Burkitt lymphoma, and one case of plasmablastic lymphoma. Ten cases (31%) were EBV negative. Of those with the morphology of diffuse large B-cell lymphoma, the EBV-negative cases were more frequently TP53-mutated (P<0.001), p53 positive by immunohistochemistry (P<0.001), CD30 negative (P<0.01), and of germinal center immunophenotype (P=0.01) compared with EBV-positive cases. No statistically significant difference in overall survival was identified based on EBV, TP53 mutation status, germinal center vs non-germinal center immunophenotype, or other immunohistochemical parameters evaluated. Patients who died of post-transplant lymphoproliferative disorder were older with a longer latency from time of transplant to diagnosis (P<0.05). Our study demonstrates that diffuse large B-cell lymphoma-related immunohistochemical prognostic markers have limited relevance in the post-transplant setting and underscores differences between EBV-positive and EBV-negative post-transplant lymphoproliferative disorder in terms of immunophenotype and TP53 mutation frequency, supporting an alternative pathogenesis for EBV-negative post-transplant lymphoproliferative disorder.
Similar content being viewed by others


Main
Post-transplant lymphoproliferative disorders are a heterogeneous group of lymphoid or plasmacytic proliferations occurring in patients with a history of solid organ or hematopoietic stem cell transplant, with a frequency of ~2% in the post-transplant population.1, 2 Monomorphic post-transplant lymphoproliferative disorders, by definition, resemble a classifiable lymphoma arising in an immunocompetent individual and are most often B-cell neoplasms, commonly diffuse large B-cell lymphoma, or Burkitt lymphoma.3
The majority of post-transplant lymphoproliferative disorders are associated with Epstein–Barr virus (EBV) infection; however, a substantial minority is not.4, 5, 6 The reported proportion of EBV-negative cases varies depending on the method used to detect EBV and the pathologic category of post-transplant lymphoproliferative disorder. EBV-negative B-cell monomorphic post-transplant lymphoproliferative disorder may represent a distinct clinicopathologic entity with a different underlying pathogenesis from EBV-positive cases. In support of this hypothesis, gene expression profiling studies have identified segregation of EBV-positive post-transplant diffuse large B-cell lymphoma from EBV-negative post-transplant diffuse large B-cell lymphoma.7 However, there is little published data regarding differences in molecular features or immunohistochemical staining patterns, beyond germinal center vs non-germinal center immunophenotype, between EBV-positive and EBV-negative cases.
Research is ongoing regarding subclassification of de novo diffuse large B-cell lymphoma arising in immunocompetent individuals into various prognostic subgroups. A common prognostic division of diffuse large B-cell lymphoma is based on the cell of origin of the tumor (germinal center B cell vs activated B cell), with the Hans immunohistochemical algorithm (germinal center vs non-germinal center immunophenotype) frequently used in clinical practice.8 Immunohistochemical expression patterns of p53, CD30, myc, and BCL2 are also potentially useful predictors of survival in de novo diffuse large B-cell lymphoma.9, 10, 11, 12, 13, 14 TP53 mutation status has been reported to have predictive value for patients with de novo diffuse large B-cell lymphoma treated with R-CHOP (rituximab, cyclophosphamide, hydroxydaunorubicin, vincristine, and prednisone) therapy.15 The potential impact of immunohistochemical prognostic markers in the context of post-transplant lymphoproliferative disorders has been reported in only a few articles.16, 17, 18
In this retrospective study from two institutions, we sought to characterize clinicopathological differences between EBV-positive and EBV-negative cases of B-cell monomorphic post-transplant lymphoproliferative disorder, including staining patterns using immunohistochemical markers often performed as part of the work-up of de novo diffuse large B-cell lymphoma, and to clarify the potential prognostic role of these markers in the post-transplant setting.
Materials and methods
Following approval by the institutional review boards of the University of Minnesota and Massachusetts General Hospital, we identified 32 cases of B-cell monomorphic post-transplant lymphoproliferative disorder (20 from the University of Minnesota by search of the electronic pathology database from 2004 through mid-2013 and 12 from Massachusetts General Hospital by search of electronic databases from 1996 through 2013). Consultation cases, cases lacking slides or blocks available for review, and cases primary to the central nervous system were excluded. Clinical information was obtained from the electronic medical records of our institutions.
Full section hematoxylin-and-eosin-stained slides and results of immunohistochemical stains performed at diagnosis were reviewed in all cases. All cases were evaluated using EBV-encoded RNA (EBER) in situ hybridization. Interpretation was obtained from the diagnostic pathology report in most cases or by interpretation of EBER in situ hybridization in a subset of cases diagnosed before routine clinical availability of EBER in situ hybridization; in the latter instance, an in situ hybridization probe from Leica Biosystems (Buffalo Grove, IL, USA) was used. A tissue microarray of the post-transplant lymphoproliferative disorder cases at Massachusetts General Hospital was manually constructed from 2 mm cores of representative areas from formalin- or B-Plus-fixed, paraffin-embedded tissue. Immunohistochemical stains for p53, myc, CD30, BCL2, CD10, BCL6, MUM1, and Ki67 were performed on the tissue microarray and on representative whole sections using techniques for routine clinical practice by the Massachusetts General Hospital and University of Minnesota immunohistochemistry laboratories, respectively (Table 1). In four cases, stains were limited to the Hans classifier (CD10, BCL6, and MUM1). In one case, the immunohistochemical interpretation was performed only on a relapse specimen, due to inadequacy of material in the initial diagnostic specimen. Interpretation of immunohistochemical stains was independently performed by at least two authors (ELC and SY or ELC and DC), with a consensus interpretation reached when needed. Detailed immunohistochemical and molecular analysis of relapse specimens was not performed.
Flow cytometry and cytogenetic analysis (karyotype and/or fluorescence in situ hybridization (FISH)) was performed using the clinical methods of the performing institution at the time, with reports reviewed. In addition, interphase FISH analysis was performed for detection of MYC gene rearrangements retrospectively on a subset of the cases with the Vysis LSI MYC Dual Color, Break Apart Rearrangement Probe (Abbott Molecular) on 4 μm sections of formalin-fixed, paraffin-embedded tissue. The assay was considered positive if the orange and green paired signals were split apart by more than two probe lengths in >15% of observed lymphoma cells (50 cell count).
TP53 sequencing was performed on formalin-fixed paraffin-embedded tissue from 27 cases with diffuse large B-cell lymphoma morphology and one case with plasmablastic morphology via amplicon-based enrichment followed by next-generation sequencing. Analysis was performed on a relapse specimen in one case as the diagnostic specimen lacked sufficient tissue. TP53 mutational analysis failed in three cases, with low-quality data precluding meaningful analysis.
Details of TP53 mutation analysis: 10 μm sections of formalin-fixed, paraffin-embedded tissue were obtained and macrodissected for tumor enrichment when necessary. DNA was extracted using the QIAamp DNA formalin-fixed, paraffin-embedded tissue kit and deparaffinization solution (Qiagen, Germantown, MD, USA) and was quantified using a Qubit 2.0 fluorometer (ThermoFisher Scientific, Waltham, MA, USA). Sequencing of TP53 was performed with amplicon-based target enrichment followed by next-generation sequencing. Amplicon enrichment was performed on the Biomark Access Array System (Fluidigm, San Francisco, CA, USA) using 12 amplicons for TP53 pooled with amplicons for targeted analysis of 20 other genes. A second step PCR reaction was performed to append barcodes and sequencing adaptors to the enriched amplicons, which were pooled and sequenced on a MiSeq instrument, version 3 chemistry (Illumina, San Diego, CA, USA).
Coverage data was analyzed for all genes to assess overall quality; however, only the variants in TP53 were analyzed. Five had suboptimal coverage but were interpretable (low coverage in non-analyzed genes but adequate coverage in TP53). Cases with a TP53 mutation and a low Qubit value (15 ng/μl or less) or suboptimal coverage were either run in duplicate (three cases) or run in duplicate and confirmed by Sanger sequencing (five cases). Wild-type TP53 cases with a low Qubit value or suboptimal coverage were run in duplicate to confirm negative results. The TP53 coding exons and five base pairs into the intron were analyzed. Known single-nucleotide polymorphisms and synonymous mutations were not considered pathogenic and these results are not shown. FASTQ files were processed through a custom designed bioinformatics pipeline for variant calling (ScanIndel version 2.0.1 (ref. 19)). Variant call files were filtered against a condensed database of non-synonymous variants of the targeted genes from the publically available COSMIC database (http://cancer.sanger.ac.uk/cosmic).20
Statistical analysis was performed using the IBM SPSS Statistics program version 22. A two-tailed Fisher’s exact test was used for categorical data and a Wilcoxon–Mann–Whitney test for equality of means was used for continuous data. Univariate survival analysis was performed using the Kaplan–Meier method with the log-rank test used to compare survival differences between groups.
Results
Clinical Features
Thirty-two cases met our inclusion criteria arising in 13 females and 19 males. The median age at diagnosis was 44 years (range 3–72). Nine patients (28%) were <18 years old and 12 patients (38%) were >50 years old at the time of diagnosis. The median length of time from the patient’s transplant (first transplant if multiple) to diagnosis of post-transplant lymphoproliferative disorder was 71 months (range 2–295). Ten cases occurred within 12 months of transplant (commonly defined as early post transplant) and five occurred greater than 12 months but <60 months after transplant. Twenty-seven of the cases occurred following solid organ transplant and five occurred following allogeneic hematopoietic stem cell transplant. The former included liver (n=9), lung (unilateral or bilateral, n=5), kidney (n=5), and heart (n=5) transplants; three had combined organ transplants (bilateral lung followed by renal transplant 10 years later, combined heart–lung, combined kidney–pancreas). Seven patients received sequential transplants of the same organ, with a range of 3–240 months between the first and last transplants; in all seven patients, the monomorphic post-transplant lymphoproliferative disorder arose after the last transplant. The indication for solid organ transplant or hematopoietic stem cell transplant varied depending on the organ transplanted and included congenital disorders, chronic diseases, and acute illness.
Diagnostic post-transplant lymphoproliferative disorder sites included lymph node (n=14), small bowel/mesentery (n=9), nasopharynx (n=2), liver (n=2), colon (n=1), anus (n=1), adrenal gland (n=1), lung (n=1), and tonsil (n=1). Post-transplant lymphoproliferative disorder diagnosed in the small bowel/mesentery were statistically more likely to be EBV negative by EBER in situ hybridization (P=0.013) and to contain a pathogenic TP53 mutation (P=0.017) than post-transplant lymphoproliferative disorder diagnosed at any other location. There was no statistically significant difference in EBV positivity or TP53 mutation status when the post-transplant lymphoproliferative disorder diagnostic site was lymph node compared with any extranodal site. Of the 30 patients with staging data, five (17%) had localized (stage 1) disease. One patient had a history of a previously diagnosed polymorphic post-transplant lymphoproliferative disorder present at a different anatomic location.
Twenty-five of the patients had peripheral blood EBV viral load obtained within 1 month of the biopsy and, of these, 10 had no increase in viral load and 15 had EBV DNA detected. On average, the EBV viral titer was drawn 0.1 months before the biopsy, with a range from 0.6 months before to 0.9 months after. Peripheral blood EBV DNA was detected in two of seven patients whose lymphoma was negative for EBV by EBER in situ hybridization. In 5 of 18 patients, peripheral blood EBV DNA was not detected; however, the lymphoma was positive for EBV by EBER in situ hybridization.
Treatment details of initial monomorphic post-transplant lymphoproliferative disorder were available for 31 patients. One received localized palliative radiation, one had a localized resection, three had a reduction in immunosuppression alone, nine received rituximab but not chemotherapy, and 17 received chemotherapy (most of the patients who received chemotherapy also received rituximab). Chemotherapy regimens included R-CHOP; bortezomib and rituximab; and rituximab, cyclophosphamide and prednisone. Two of three patients diagnosed with Burkitt lymphoma were treated with Burkitt lymphoma-directed treatment protocols; one was treated with rituximab and methylprednisolone only (the patient died before more aggressive therapy could be initiated).
Five cases, all with diffuse large B-cell lymphoma morphology and EBV positive, arose after hematopoietic stem cell transplant. In contrast to solid organ transplant patients, most patients with a history of hematopoietic stem cell transplant were <18 years old (4/5, P=0.015), none received chemotherapy (P=0.012), all occurred within 12 months of transplant (P=0.001), and none were p53 positive (P=0.041). One patient had a TP53 mutation. All patients received rituximab. At last follow-up (median follow-up of 22 months, range 6–108), four hematopoietic stem cell transplant patients were alive and one was deceased from a cause other than post-transplant lymphoproliferative disorder.
Morphology and Immunohistochemistry
Morphology of the monomorphic post-transplant lymphoproliferative disorder included 27 cases with the morphology of diffuse large B-cell lymphoma, three with the morphology of Burkitt lymphoma, one case of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma, and one with the morphology of plasmablastic lymphoma. For the purpose of our analysis, the case of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma was included with the diffuse large B-cell lymphoma cases. Three were reclassified from an original diagnosis of polymorphic post-transplant lymphoproliferative disorder to monomorphic post-transplant lymphoproliferative disorder based on 2008 WHO criteria2 and consensus review by study authors. B-cell lineage was confirmed in all cases by immunohistochemical staining and/or flow cytometric immunophenotyping. On the basis of immunohistochemical stains performed at the time of diagnosis, the plasmablastic lymphoma was positive for CD79a (weak), CD138 and MUM1, and negative for CD20, Pax5, T-cell markers and human herpes virus-8; the case was EBV-positive by EBER.
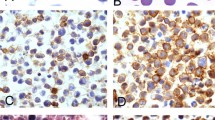
Additional immunohistochemical staining results evaluated for this study are shown in Table 2. Twenty-two cases (69%) were EBV positive and 10 (31%) were EBV negative. In the EBV-positive cases, EBV was positive in the majority of neoplastic cells with one exception (a single case with the morphology of Burkitt lymphoma that had scattered EBV-positive cells). Table 3 compares the clinicopathologic features of EBV-positive and EBV-negative monomorphic post-transplant lymphoproliferative disorder cases. In terms of immunohistochemical features, EBV-negative cases were more frequently CD30-negative (P=0.003), p53-positive (P=0.004), and of germinal center immunophenotype (P=0.01) compared with EBV-positive cases (Figure 1). These statistically significant differences in immunohistochemical pattern persisted after exclusion of the five cases occurring following hematopoietic stem cell transplant.

(a–c) Representative images from Case 20, Epstein–Barr virus (EBV)-positive monomorphic post-transplant lymphoproliferative disorder (diffuse large B-cell lymphoma): (a) hematoxylin and eosin, 40 × objective; (b) MUM1 immunohistochemical stain, 10 × objective; (c) EBV RNA in situ hybridization (EBER), 10 × objective. (d–f) Representative images from Case 12, EBV-negative monomorphic post-transplant lymphoproliferative disorder (diffuse large B-cell lymphoma): (d) hematoxylin and eosin, 40 × objective; (e) CD10 immunohistochemical stain, 10 × objective; (f) p53 immunohistochemical stain, 10 × objective.
The three cases with Burkitt lymphoma morphology occurred after solid organ transplant, had disease at multiple other sites, and had detectable EBV DNA in the peripheral blood. Two patients were adults (ages 25 and 46 years) and one was a child (9 years old) at the time of diagnosis. The post-transplant lymphoproliferative disorder occurred ~132 months after transplant in the two adults, and 48 months after transplant in the child. At last follow-up, one patient was alive, one had died of post-transplant lymphoproliferative disorder, and one was deceased from a cause other than post-transplant lymphoproliferative disorder. None of the cases were positive for BCL2 or CD30 by immunohistochemistry and all were positive for EBV by in situ hybridization.
TP53 Mutational Analysis
Eleven of 25 evaluable cases (44%) had a pathogenic TP53 mutation (Table 4). All 11 positive cases had the morphology of diffuse large B-cell lymphoma. The single case of plasmablastic lymphoma had wild-type TP53. TP53 sequencing was not performed for the monomorphic post-transplant lymphoproliferative disorder cases with Burkitt lymphoma morphology. Of the 11 cases with a pathogenic TP53 mutation, four cases had two mutations and one had three mutations. A total of 17 mutations were identified, all missense mutations except one nonsense mutation. Mutations occurred most commonly in exons 7 and 8, with seven and six mutations, respectively. Codons 231, 248 and 273 were mutated in more than one patient. Mutations were more commonly G>A (6 mutations), C>T (3 mutations), or A>G (3 mutations). Comparison of next-generation sequencing replicates and Sanger confirmation was used to ensure low level C>T/G>A mutations were not due to deamination (fixation) artifact.
Eight of the 11 cases with a TP53 mutation (73%) were positive for p53 by immunohistochemistry, with staining of greater than 30% of neoplastic cells (Table 4). Two of the 14 cases with wild-type TP53 (14%) were positive for p53 by immunohistochemistry, with staining of 40–50% of neoplastic cells. As shown in Table 3, the majority of TP53-mutated monomorphic post-transplant lymphoproliferative disorder diffuse large B-cell lymphoma cases (9/11, 82%) were EBV negative, with all nine EBV-negative cases harboring a TP53 mutation vs only 2/15 EBV-positive cases (13%, P<0.001).
Flow Cytometry and Cytogenetics
Twenty-three cases had concurrent immunophenotyping by flow cytometry. Twelve (52%) had an identifiable monotypic B-cell population (four lambda restricted, eight kappa restricted), whereas five had abnormal B cells lacking surface or cytoplasmic light chain expression. One case with diffuse large B-cell lymphoma morphology had lambda monotypic plasma cells and polytypic B cells.
Eighteen cases had cytogenetic evaluation by karyotype and/or FISH (Table 5). MYC rearrangements were identified by break-apart probe in two of the twelve evaluated cases, both with the morphology of Burkitt lymphoma. No BCL6 or BCL2 rearrangements were detected by FISH (evaluated in five cases). Karyotype was obtained in ten cases and was abnormal in six, including one (case 31) with the morphology of B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma that had an unbalanced three-way rearrangement involving MYC, IGH and an unknown partner on the short arm of chromosome 4.
Outcome and Survival Analysis
At last follow-up (median 44 months for surviving patients, range 4–214), 18 patients were alive without evidence of post-transplant lymphoproliferative disorder, one was alive with post-transplant lymphoproliferative disorder, seven died of post-transplant lymphoproliferative disorder, and six died of another cause. Details of patients deceased of post-transplant lymphoproliferative disorder in comparison to those with other outcomes are shown in Table 6. Nine patients relapsed with monomorphic post-transplant lymphoproliferative disorder of a similar morphology a median of 14 months (range 2–55) after first monomorphic post-transplant lymphoproliferative disorder diagnosis. Of the seven patients who died of post-transplant lymphoproliferative disorder, five died after disease recurrence, with date of death ranging from less than a month to almost 12 months after relapse. Two of 11 patients (18%) with TP53-mutated monomorphic post-transplant lymphoproliferative disorder died of post-transplant lymphoproliferative disorder. Similarly, 3 of 14 patients (21%) with TP53 wild-type monomorphic post-transplant lymphoproliferative disorder died of post-transplant lymphoproliferative disorder.
The various immunohistochemical/in situ hybridization parameters and clinical factors evaluated (as listed in Tables 2 and 6) showed no significant association with overall survival, as defined as the time from diagnosis to death from any cause. There was no significant association with TP53 mutation status and overall survival for all cases and when analysis was restricted to cases with diffuse large B-cell lymphoma morphology or EBV negativity. When analysis was restricted to case with diffuse large B-cell lymphoma morphology, there was a trend toward shorter overall survival among patients who relapsed (P=0.078).
Discussion
We performed an extensive clinicopathologic analysis of B-cell monomorphic post-transplant lymphoproliferative disorder, most with the morphology of diffuse large B-cell lymphoma, and evaluated for TP53 mutations. We found EBV-negative cases to be more frequently TP53-mutated and p53-positive, CD30-negative and of germinal center type by immunohistochemistry compared with EBV-positive cases.
EBV-negative monomorphic post-transplant lymphoproliferative disorder may arise through different pathogenic mechanisms and more closely resemble lymphomas arising in immunocompetent hosts than EBV-positive monomorphic post-transplant lymphoproliferative disorder. In this study, we identified statistically significant differences in TP53 mutation frequency (P<0.001) and p53 expression by immunohistochemistry (P<0.001) in EBV-negative vs EBV-positive cases with diffuse large B-cell lymphoma morphology, supporting the distinctive nature of EBV-negative monomorphic post-transplant lymphoproliferative disorder and pointing toward a probable mechanism involving p53 in its pathogenesis. TP53 mutations have a reported frequency of 13–22% in de novo diffuse large B-cell lymphoma, and p53 overexpression has been reported in 20–60% of cases.15, 21, 22, 23, 24 In a study of 22 cases of post-transplant lymphoproliferative disorder that included both polymorphic and monomorphic types, 16 had at least some p53 expression (>1% of cells positive), whereas two had high p53 expression (>75% of cells positive).16 Half of the cases in our series were positive for p53 with greater than 30% of cells positive. Few cases of post-transplant lymphoproliferative disorder have been evaluated for TP53 mutations.25, 26 Previously reported mutations include a frameshift mutation in codon 301 and a missense mutation in codon 248 leading to an amino-acid change from arginine to leucine. In our series, exons 5–8 were most often mutated and the majority of mutations (94%) occurred in the DNA-binding domain (residues 96–292), similar to findings reported in de novo diffuse large B-cell lymphoma.15, 21, 22, 27 Six mutations in our series (35%) occurred at known ‘hotspot’ locations (codons 175, 248, and 273) for mature B-cell lymphomas.28
Other genetic changes reported in post-transplant lymphoproliferative disorder include alterations of MYC and BCL6, DNA hypermethylation, aberrant somatic hypermutation, and microsatellite instability.26, 29, 30, 31, 32, 33, 34 Case 27 in our series had both a TP53 mutation and an abnormal karyotype showing loss of the entire short arm of chromosome 17, resulting in TP53 loss of heterozygosity. In our series, the only recurrent cytogenetic abnormality was a MYC rearrangement in three cases (two EBV-positive Burkitt lymphoma and one EBV-negative B-cell lymphoma, unclassifiable, with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma). Given the frequency of MYC translocations in de novo diffuse large B-cell lymphoma and the implications of a ‘double-hit’ lymphoma diagnosis in immunocompetent individuals,35, 36 we suggest further studies of monomorphic post-transplant lymphoproliferative disorder include FISH evaluation for MYC, BCL2, and BCL6 translocations.
The frequency of EBV-negative cases in this series (31%) is similar to that reported previously in studies focused on B-cell monomorphic post-transplant lymphoproliferative disorder.17, 18 The EBV-negative cases in our series had a longer latency, were more likely to be diagnosed in the small bowel/mesentery, and were more likely to have a germinal center immunophenotype compared with EBV-positive cases. The latter result supports the association of a non-germinal center immunophenotype with EBV positivity,6, 37 a finding that may be related to the reservoir of latent EBV infection (long-lived memory B cells).38 The association of EBV-negative cases with diagnosis in the small bowel/mesentery may imply a unique aspect of the immune microenvironment in that location; further investigation with more cases is required. The EBV-positive cases occurring in patients <18 years of age and within a year of transplant were disproportionately from patients with a history of hematopoietic stem cell transplant. We found no significant survival difference between EBV-positive and EBV-negative monomorphic post-transplant lymphoproliferative disorder, similar to previously reported studies.17, 18
In a subset of the cases in our series, there was a discrepancy between peripheral blood EBV viral load and EBV by EBER in situ hybridization of the monomorphic post-transplant lymphoproliferative disorder tissue. Technical failure (for example, due to poor RNA quality) or levels below the limit of detection of the assay are possible reasons for this discrepancy. Generalizations regarding technique are difficult as our cases included two institutions and spanned more than a decade. We used the qualitative interpretation of EBV DNA levels, with variable interpretive cutoffs depending on the institution and year. We documented EBV viral loads within 1 month before or after the tissue biopsy. In some cases, the discrepancy could be due to the length of time between the peripheral blood viral load evaluation and procurement of the biopsy specimen and the effects of any intervening therapy. In addition, localization of the EBV-driven process to the neoplastic lymphatic tissue without peripheral blood circulation is a possibility.
De novo diffuse large B-cell lymphoma is routinely subclassified for prognostic purposes into germinal center and non-germinal center types based on immunohistochemical algorithms, such as the Hans classifier, with a better overall survival in the germinal center group.8, 39 This division serves as a surrogate for gene expression profiling-based classification, in which tumor cell of origin from germinal center B cells is associated with longer overall survival than origin from activated B cells.40, 41 No significant difference in outcome has been shown for germinal center vs non-germinal center monomorphic post-transplant lymphoproliferative disorder by immunophenotyping,17, 18 similar to our findings. The recent availability of RNA-based expression platforms designed for use on formalin-fixed paraffin-embedded tissue should allow for more accurate cell of origin classification in the near future and enable further confirmation of the lack of prognostic relevance of cell of origin in monomorphic post-transplant lymphoproliferative disorder.42
Immunohistochemical prognostic markers beyond cell of origin have not been rigorously studied in monomorphic post-transplant lymphoproliferative disorder. Other potentially useful predictors of survival in de novo diffuse large B-cell lymphoma include immunohistochemical expression patterns of p53, CD30, myc, and BCL2 and the presence or absence of TP53 mutations.9, 10, 11, 12, 13, 14, 15, 43 We found no statistically significant difference in outcome for any of these immunohistochemical parameters or for TP53 mutation status, even when analysis was restricted to EBV-negative or diffuse large B-cell lymphoma cases. However, given the small number of patients studied and the non-uniform therapy given, further study is needed to confirm our findings.
A variety of clinical factors have been identified as poor prognostic indicators in post-transplant lymphoproliferative disorder, including increased age, elevated serum lactate dehydrogenase, low serum albumin, presence of B-symptoms, advanced stage disease and central nervous system involvement.44, 45 Patients with primary central nervous system disease were excluded from this study as primary central nervous system post-transplant lymphoproliferative disorder may represent a distinct entity from systemic post-transplant lymphoproliferative disorder and is usually treated differently.44, 46 Clinical parameters evaluated in our study included age, disease stage, and latency (time from transplant to monomorphic post-transplant lymphoproliferative disorder diagnosis). No survival advantage was seen in patients with localized disease. Patients who died of post-transplant lymphoproliferative disorder were older at the time of diagnosis and had a longer latency, suggesting these two factors are indicators of poorer prognosis in our series.
Five patients in our series developed monomorphic post-transplant lymphoproliferative disorder following allogeneic hematopoietic stem cell transplant. These patients shared similar characteristics to previously reported hematopoietic stem cell transplant patients with post-transplant lymphoproliferative disorder,47, 48 with all but one under the age of 18, all occurring within 12 months of transplant, and all with a non-germinal center immunophenotype. Risk factors for post-transplant lymphoproliferative disorder following hematopoietic stem cell transplant differ from those following solid organ transplant, and include antithymocyte globulin use and features of the stem cell source including umbilical cord blood transplantation, unrelated donor/HLA-mismatched donors, and T-cell depletion of donor marrow.44, 48, 49 All of the hematopoietic stem cell transplant patients in our series had at least one, and frequently multiple, of these risk factors (data not shown).
In summary, our study highlights the distinctive nature of EBV-negative B-cell monomorphic post-transplant lymphoproliferative disorder with the novel finding of TP53 mutations in all EBV-negative cases with diffuse large B-cell lymphoma morphology, pointing to a role for TP53 in their pathogenesis. No difference was seen in overall survival based on germinal center vs non-germinal center immunophenotype or the immunohistochemical expression of p53, CD30, myc, or BCL2, suggesting that immunohistochemical prognostic markers important in de novo diffuse large B-cell lymphoma do not apply to the post-transplant setting. Survival findings require validation in larger series of uniformly treated patients with B-cell monomorphic post-transplant lymphoproliferative disorder.
References
Al-Mansour Z, Nelson BP, Evens AM . Post-transplant lymphoproliferative disease (PTLD): risk factors, diagnosis, and current treatment strategies. Curr Hematol Malig Rep 2013;8:173–183.
Swerdlow SH, Webber SA, Chadburn A et al. Post-transplant lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL et al (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC: Lyon, France, 2008, pp 343–349.
Bagg A, Dunphy CH . Immunosuppressive and immunomodulatory therapy-associated lymphoproliferative disorders. Semin Diagn Pathol 2013;30:102–112.
Nelson BP, Nalesnik MA, Bahler DW et al. Epstein-Barr virus-negative post-transplant lymphoproliferative disorders: a distinct entity? Am J Surg Pathol 2000;24:375–385.
Leblond V, Davi F, Charlotte F et al. Posttransplant lymphoproliferative disorders not associated with Epstein-Barr virus: a distinct entity? J Clin Oncol 1998;16:2052–2059.
Capello D, Cerri M, Muti G et al. Molecular histogenesis of posttransplantation lymphoproliferative disorders. Blood 2003;102:3775–3785.
Morscio J, Dierickx D, Ferreiro JF et al. Gene expression profiling reveals clear differences between EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am J Transplant 2013;13:1305–1316.
Hans CP, Weisenburger DD, Greiner TC et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004;103:275–282.
Xie Y, Bulbul MA, Ji L et al. p53 expression is a strong marker of inferior survival in de novo diffuse large B-cell lymphoma and may have enhanced negative effect with MYC coexpression: a single institutional clinicopathologic study. Am J Clin Pathol 2014;141:593–604.
Hu S, Xu-Monette ZY, Balasubramanyam A et al. CD30 expression defines a novel subgroup of diffuse large B-cell lymphoma with favorable prognosis and distinct gene expression signature: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2013;121:2715–2724.
Hu S, Xu-Monette ZY, Tzankov A et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013;121:4021–4031; quiz 250.
Horn H, Ziepert M, Becher C et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013;121:2253–2263.
Johnson NA, Slack GW, Savage KJ et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 2012;30:3452–3459.
Iqbal J, Meyer PN, Smith LM et al. BCL2 predicts survival in germinal center B-cell-like diffuse large B-cell lymphoma treated with CHOP-like therapy and rituximab. Clin Cancer Res 2011;17:7785–7795.
Xu-Monette ZY, Wu L, Visco C et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012;120:3986–3996.
Oton AB, Wang H, Leleu X et al. Clinical and pathological prognostic markers for survival in adult patients with post-transplant lymphoproliferative disorders in solid transplant. Leuk Lymphoma 2008;49:1738–1744.
Johnson LR, Nalesnik MA, Swerdlow SH . Impact of Epstein-Barr virus in monomorphic B-cell posttransplant lymphoproliferative disorders: a histogenetic study. Am J Surg Pathol 2006;30:1604–1612.
Kinch A, Baecklund E, Backlin C et al. A population-based study of 135 lymphomas after solid organ transplantation: the role of Epstein-Barr virus, hepatitis C and diffuse large B-cell lymphoma subtype in clinical presentation and survival. Acta Oncol 2014;53:669–679.
Yang R, Nelson AC, Henzler C et al. ScanIndel: a hybrid framework for indel detection via gapped alignment, split reads and de novo assembly. Genome Med 2015;7:127.
Forbes SA, Beare D, Gunasekaran P et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811.
Young KH, Leroy K, Moller MB et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood 2008;112:3088–3098.
Young KH, Weisenburger DD, Dave BJ et al. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood 2007;110:4396–4405.
Paik JH, Jeon YK, Park SS et al. Expression and prognostic implications of cell cycle regulatory molecules, p16, p21, p27, p14 and p53 in germinal centre and non-germinal centre B-like diffuse large B-cell lymphomas. Histopathology 2005;47:281–291.
Chang CC, Liu YC, Cleveland RP, et al. Expression of c-Myc and p53 correlates with clinical outcome in diffuse large B-cell lymphomas. Am J Clin Pathol 2000;113:512–518.
Knowles DM, Cesarman E, Chadburn A et al. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood 1995;85:552–565.
Capello D, Rossi D, Gaidano G . Post-transplant lymphoproliferative disorders: molecular basis of disease histogenesis and pathogenesis. Hematol Oncol 2005;23:61–67.
Martin AC, Facchiano AM, Cuff AL et al. Integrating mutation data and structural analysis of the TP53 tumor-suppressor protein. Hum Mutat 2002;19:149–164.
Petitjean A, Mathe E, Kato S et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007;28:622–629.
Capello D, Gaidano G . Post-transplant lymphoproliferative disorders: role of viral infection, genetic lesions and antigen stimulation in the pathogenesis of the disease. Mediterr J Hematol Infect Dis 2009;1:e2009018.
Duval A, Raphael M, Brennetot C et al. The mutator pathway is a feature of immunodeficiency-related lymphomas. Proc Natl Acad Sci USA 2004;101:5002–5007.
Delecluse HJ, Rouault JP, Jeammot B et al. Bcl6/Laz3 rearrangements in post-transplant lymphoproliferative disorders. Br J Haematol 1995;91:101–103.
Cesarman E, Chadburn A, Liu YF et al. BCL-6 gene mutations in posttransplantation lymphoproliferative disorders predict response to therapy and clinical outcome. Blood 1998;92:2294–2302.
Rossi D, Gaidano G, Gloghini A et al. Frequent aberrant promoter hypermethylation of O6-methylguanine-DNA methyltransferase and death-associated protein kinase genes in immunodeficiency-related lymphomas. Br J Haematol 2003;123:475–478.
Capello D, Rasi S, Oreste P et al. Molecular characterization of post-transplant lymphoproliferative disorders of donor origin occurring in liver transplant recipients. J Pathol 2009;218:478–486.
Petrich AM, Nabhan C, Smith SM . MYC-associated and double-hit lymphomas: A review of pathobiology, prognosis, and therapeutic approaches. Cancer 2014;120:3884–3895.
Barrans S, Crouch S, Smith A et al. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol 2010;28:3360–3365.
Capello D, Berra E, Cerri M et al. Post-transplant lymphoproliferative disorders. Molecular analysis of histogenesis and pathogenesis. Minerva Med 2004;95:53–64.
Nourse JP, Jones K, Gandhi MK . Epstein-Barr Virus-related post-transplant lymphoproliferative disorders: pathogenetic insights for targeted therapy. Am J Transplant 2011;11:888–895.
Meyer PN, Fu K, Greiner TC et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J Clin Oncol 2011;29:200–207.
Lenz G, Wright G, Dave SS et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008;359:2313–2323.
Alizadeh AA, Eisen MB, Davis RE et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–511.
Scott DW, Wright GW, Williams PM et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014;123:1214–1217.
Iqbal J, Neppalli VT, Wright G et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J Clin Oncol 2006;24:961–968.
Singavi AK, Harrington AM, Fenske TS . Post-transplant lymphoproliferative disorders. Cancer Treat Res 2015;165:305–327.
Jagadeesh D, Woda BA, Draper J, et al. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options Oncol 2012;13:122–136.
Evens AM, Choquet S, Kroll-Desrosiers AR et al. Primary CNS posttransplant lymphoproliferative disease (PTLD): an international report of 84 cases in the modern era. Am J Transplant 2013;13:1512–1522.
Novoa-Takara L, Perkins SL, Qi D et al. Histogenetic phenotypes of B cells in posttransplant lymphoproliferative disorders by immunohistochemical analysis correlate with transplant type: solid organ vs hematopoietic stem cell transplantation. Am J Clin Pathol 2005;123:104–112.
Rasche L, Kapp M, Einsele H et al. EBV-induced post transplant lymphoproliferative disorders: a persisting challenge in allogeneic hematopoetic SCT. Bone Marrow Transplant 2014;49:163–167.
Landgren O, Gilbert ES, Rizzo JD et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 2009;113:4992–5001.
Acknowledgements
We thank Matthew Schomaker and Aaron Lambert (Molecular Diagnostics Laboratory, University of Minnesota Health); Karin Daniels and Dr. Monna Marolt (Immunohistochemistry Laboratory, University of Minnesota Health); Dr. Paola Dal Cin (Brigham and Women’s Hospital); and Arthur Karakatsanis (Histology Laboratory, Massachusetts General Hospital) for their assistance with this project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Courville, E., Yohe, S., Chou, D. et al. EBV-negative monomorphic B-cell post-transplant lymphoproliferative disorders are pathologically distinct from EBV-positive cases and frequently contain TP53 mutations. Mod Pathol 29, 1200–1211 (2016). https://doi.org/10.1038/modpathol.2016.130
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.130
This article is cited by
-
Important Considerations in the Diagnosis and Management of Post-transplant Lymphoproliferative Disorder
Current Oncology Reports (2023)
-
Targeted massively parallel sequencing of mature lymphoid neoplasms: assessment of empirical application and diagnostic utility in routine clinical practice
Modern Pathology (2021)
-
Post-transplantation lymphoproliferative disorder after haematopoietic stem cell transplantation
Annals of Hematology (2021)
-
Joining Efforts for PTLD: Lessons Learned from Comparing the Approach and Treatment Strategies Across the Pediatric and Adult Age Spectra
Current Hematologic Malignancy Reports (2021)
-
Second Malignancies after Hematopoietic Stem Cell Transplantation
Current Treatment Options in Oncology (2018)
