
Abstract
Germline deletions affecting the Epithelial cell adhesion molecule (EPCAM) gene lead to silencing of MSH2 and cause Lynch syndrome. We have recently reported that lack of EPCAM expression occurs in many, but not all tumors from Lynch syndrome patients with EPCAM germline deletions. The differences in EPCAM expression were not related to the localization of EPCAM germline deletions. We therefore hypothesized that the type of the second somatic hit, which leads to MSH2 inactivation during tumor development, determines EPCAM expression in the tumor cells. To test this hypothesis and to evaluate whether lack of EPCAM expression can already be detected in Lynch syndrome-associated adenomas, we analyzed four carcinomas and two adenomas from EPCAM germline deletion carriers for EPCAM protein expression and allelic deletion status of the EPCAM gene region by multiplex ligation-dependent probe amplification. In four out of six tumors we observed lack of EPCAM expression accompanied by biallelic deletions affecting the EPCAM gene. In contrast, monoallelic retention of the EPCAM gene was observed in the remaining two tumors with retained EPCAM protein expression. These results demonstrate that EPCAM expression in tumors from EPCAM deletion carriers depends on the localization of the second somatic hit that inactivates MSH2. Moreover, we report lack of EPCAM protein expression in a colorectal adenoma, suggesting that EPCAM immunohistochemistry may detect EPCAM germline deletions already at a precancerous stage.
Similar content being viewed by others

Main
Lynch syndrome, clinically referred to as hereditary non-polyposis colorectal cancer (HNPCC), is one of the most common hereditary cancer predisposition syndromes that is associated with a high-lifetime risk of colorectal carcinomas and extracolonic tumors including endometrial cancers. Lynch syndrome is responsible for about 3% of colorectal cancers.1 Germline mutations underlying Lynch syndrome affect the DNA mismatch repair genes2 such as MLH13 or MSH2,4 and less frequently MSH65 or PMS2.6 Lynch syndrome-associated tumors are characterized by DNA mismatch repair deficiency, which results from a second somatic event inactivating the respective remaining functional mismatch repair gene allele.7 As a consequence, tumorigenesis is promoted by secondary mutations that accumulate at short repetitive sequences, a phenotype termed high-level microsatellite instability.
In Lynch syndrome diagnostics, the detection of high-level microsatellite instability in tumor tissue is supplemented by mismatch repair protein immunohistochemistry, which allows predicting the mismatch repair gene affected by a germline mutation.8
Deletions affecting the polyadenylation site located in exon 9 of the Epithelial cell adhesion molecule (EPCAM) gene located upstream of MSH2 have recently been identified as a novel cause of Lynch syndrome.9 EPCAM deletions lead to a transcriptional read-through, silencing MSH29 and are estimated to cause Lynch syndrome in ∼20–25% of patients with MSH2-negative cancers, but no detectable MSH2 germline mutation.10 This corresponds to ∼2–3% of Lynch syndrome patients.10
A recent study demonstrated that concomitant lack of EPCAM and MSH2 protein expression is a feature highly specific for cancers from EPCAM deletion carriers, suggesting EPCAM immunohistochemistry as a potential tool for the identification of Lynch syndrome patients with EPCAM germline deletions.11 However, EPCAM protein expression was retained in some cancers from EPCAM deletion carriers. There was no relation between EPCAM protein expression status in cancers and the localization of the EPCAM germline deletion.11
We here hypothesized that the localization of the second somatic hit leading to MSH2 inactivation determined EPCAM expression in the tumor cells. To that end, we analyzed EPCAM protein expression and allelic deletion status in the EPCAM gene by multiplex ligation-dependent probe amplification (MLPA) using DNA isolated from tumor tissue and corresponding blood samples in EPCAM germline deletion carriers.
Materials and methods
Patients' Characteristics and Tumors
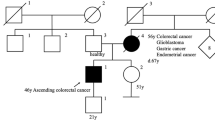
Clinical specimens used in this study were collected at the Department of Applied Tumor Biology, Institute of Pathology, University Hospital Heidelberg in the context of the German HNPCC Consortium funded by the Deutsche Krebshilfe (German Cancer Aid). Tumors and corresponding blood samples were obtained from five Lynch syndrome patients with known germline deletions affecting a genetic region encompassing exon 9 of the EPCAM gene. Family history and clinicopathological data are provided in Figure 1. Patients 1 and 2 were related, sharing the same germline deletion, and patients 3, 4, and 5 were from independent families. Exon-wise sequencing of the MLH1 and MSH2 genes had been performed in all index patients (patients 1, 3, 4, and 5), and no pathogenic germ line mutations were detected in these genes. Results of EPCAM immunohistochemistry of tumors from patients 3, 4, and 5 (colorectal carcinomas) and patient 1 (small bowel carcinoma) have been presented previously.11 In addition, two colorectal adenomas (patient 1 and 2) were included in the present study.

Family history of included patients (a–d). Index patients are marked by arrows (P1–P5), analyzed tumors are underlined and bold (T1–T6). Clinical and histopathology information is provided in (e). Tumors are abbreviated as follows: Adenoma, colorectal adenoma; CRC, colorectal cancer; GC, gastric cancer; NHL, non-Hodgkin lymphoma.
The study was approved by the Institutional Ethics Committee, and written informed consent was obtained from all patients included in this study.
DNA Isolation
DNA was extracted from whole blood and tumor specimens using the Qiagen DNeasy blood and tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Immunohistochemistry
Immunohistochemistry of MSH2 and EPCAM was performed using mouse monoclonal antibody clones FE11 (dilution 1:200, Calbiochem, Gibbstown, NJ, USA) and BER-Ep4 (1:100, Dako, Glostrup, Denmark), respectively, following standard procedures. Briefly, tissue sections (2 μm) from formalin-fixed paraffin embedded tissue were mounted on aminopropylsilane-coated slides (SuperFrost, Menzel, Braunschweig, Germany). Following deparaffinization and rehydration, slides were boiled in 10 mM citrate buffer (pH 6) for 15 min for antigen retrieval. After cooling for 20 min, endogenous peroxidase activity was blocked by incubation with 0.6% H2O2 in methanol during 20 min. Sections were blocked with 10% normal horse serum (Vectastain Elite ABC kit, Vector, Burlingame, CA, USA), before the application of the primary antibodies, which was followed by incubation over night at 4°C. After washing, biotinylated secondary antibody (1:50 dilution, horse anti-mouse IgG, Vectastain Elite ABC kit, Vector) was applied, followed by incubation at room temperature for 30 min, and the application of the AB reagent according to the manufacturer's instructions (Vectastain Elite ABC kit). Visualization was performed using AEC chromogen (Dako), and sections were counterstained with hematoxylin (AppliChem, Darmstadt, Germany) and mounted with Aquatex (Merck, Darmstadt, Germany).
Multiplex Ligation-Dependent Probe Amplification
MLPA is a method for the detection of genomic deletions by quantitative hybridization and subsequent multiplex amplification of probes specific for different regions of a gene or multiple genes of interest.12 Here, MLPA was used to detect deletions affecting the EPCAM and/or MSH2 genes.
We performed MLPA by using the SALSA MLPA kits P072-B1 and P003-B1 (MRC Holland, Amsterdam, The Netherlands), following the manufacturer's instructions. The kits contain probes for sequences within exons 3, 8, and 9 of EPCAM, exons 1–16 of MSH2 and three probes for sequences in the intergenic region between EPCAM and MSH2. MLPA was performed using DNA isolated from formalin-fixed tumor tissues to evaluate potential somatic deletions. DNA isolated from corresponding blood samples was used to quantify the germline allelic status. DNA of a healthy donor who did not harbor any deletions affecting the genomic region encompassing EPCAM and MSH2 was used as reference.
MLPA analysis was performed twice for each sample. Amplified products were visualized on an ABI3100 sequencer (Applied Biosystems, Darmstadt, Germany). Peak areas were quantified according to the manufacturer's suggestions for intra-sample and inter-sample normalization. Briefly, peak areas of each probe were divided by the sum of the reference probe peak areas for each of the samples separately to ensure intra-sample normalization. Intra-normalized probe ratios were then divided by intra-normalized probe ratios of the reference sample. Ratios lower than 0.8 were regarded as heterozygous deletions, lower than 0.3 as biallelic deletions.
Exon-wise sequencing of MSH2 was performed for the colorectal adenoma from patient 1 as described previously.13
Results
EPCAM Protein Expression
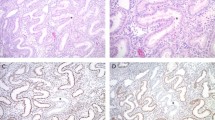
EPCAM protein expression was absent in four out of six tumors (two out of three CRCs, one small bowel cancer, one out of two colorectal adenomas) from heterozygous EPCAM germline deletion carriers and retained in the remaining two tumors. EPCAM loss was spatially congruent with MSH2 loss in all specimens. Representative staining results of the colorectal adenoma from patient 1 are shown in Figure 2.

Concomitant loss of EPCAM (a) and MSH2 (b) protein expression is observed in a colorectal adenoma from an EPCAM germline deletion carrier with Lynch syndrome (patient 2, × 100 magnification). EPCAM loss colocalized with MSH2 loss (arrows), suggesting synchronous inactivation of EPCAM and MSH2 through the same somatic event. The adenoma from patient 1 ( × 100 magnification, lower panel) shows lack of MSH2 (c), but retained EPCAM expression (d).
Deletions of EPCAM and EPCAM Protein Expression
MLPA analysis from tumor tissue revealed biallelic deletions affecting the EPCAM gene in four out of six analyzed tumors. In the remaining two tumors, no biallelic EPCAM deletions were detected, and the allelic profile obtained for the EPCAM gene region was identical in DNA isolated from tumor tissue and matched blood samples.
All tumors showing biallelic deletions in the EPCAM gene region were negative for EPCAM protein expression, whereas EPCAM protein expression was retained in the tumors with a retained second EPCAM allele (CRC from patient 5 and colorectal adenoma from patient 1, Figure 3). In one of these tumors (colorectal adenoma from patient 1), the heterozygous mutation c.942+3A>T, which is known to inactivate MSH2,14 was identified as the second somatic hit causing loss of MSH2.

Results of EPCAM deletion analysis. MLPA probes are shown in relation to the genomic region encompassing EPCAM and the 5′ region of MSH2. MLPA probes are denoted by fragment lengths (SALSA MLPA kit P072-B1, MRC Holland). White circles: biallelic presence, gray circles: heterozygous deletion, black circles: biallelic deletion. a—Exons 1–15 of the MSH2 gene showed heterozygous deletion in blood and biallelic deletion in the tumor, exon 16 was biallelically retained in blood and tumor.
In three out of four tumors, germline deletions were exactly mirrored by the corresponding somatic deletions (Figure 3, tumors from patient 1, 2, and 3). In one tumor (tumor from patient 4) the somatic deletion affecting EPCAM spanned a smaller area than the germline deletion (Figure 3).
Discussion
Concomitant loss of EPCAM and MSH2 protein expression is a typical feature of tumors from EPCAM germline deletion carriers with Lynch syndrome.11 In this study, we correlated EPCAM protein expression with the second somatic hit that leads to the inactivation of MSH2 in Lynch syndrome patients. Only if the second hit affected the EPCAM gene resulting in a biallelic EPCAM deletion, we observed a loss of EPCAM protein expression. EPCAM loss was spatially congruent with the loss of MSH2 protein expression in all analyzed lesions (Figure 2), suggesting that one somatic event led to the simultaneous inactivation of both EPCAM and MSH2.
In tumors with retained EPCAM expression, the MLPA profile obtained for the EPCAM gene region was identical to the profile of corresponding normal blood cells. These data demonstrate that the lack of EPCAM protein expression observed in tumors from EPCAM deletion carriers requires biallelic EPCAM deletion, which results from a combination of germline alteration and second somatic hit. This, vice versa, implies that heterozygous EPCAM germline deletions are not necessarily associated with loss of EPCAM expression in tumor tissue. The detection of a somatic mutational event causing MSH2 inactivation in one of the EPCAM-positive tumors (adenoma from patient 1) explains why some tumors from EPCAM germline deletion carriers show loss of MSH2, but retained EPCAM expression.11
The crucial role of the second hit in determining EPCAM protein expression is further illustrated by the comparative analysis of three tumors from two related individuals who shared the same EPCAM deletion (patients 1 and 2, Figure 3). Consistent with EPCAM protein expression status, EPCAM MLPA revealed biallelic deletions affecting the EPCAM gene in the EPCAM-negative tumors, but monoallelic retention of EPCAM in the EPCAM-positive adenoma.
EPCAM immunohistochemistry has been suggested as a potential diagnostic tool in Lynch syndrome.11 However, the dependence of EPCAM expression on both, germline and somatic alterations explains why EPCAM immunohistochemistry can yield inconspicuous results in a subset of tumors from EPCAM deletion carriers, namely if the second somatic MSH2-inactivating event does not affect the EPCAM gene.
The frequent occurrence of somatic deletions affecting the EPCAM gene as a second hit in tumors from EPCAM deletion carriers suggests that the localization of somatic events inactivating mismatch repair genes in Lynch syndrome is not random, but related to the underlying germline mutation. This is in line with the previous observation that large deletions affecting the MLH1 or MSH2 gene are particularly frequent in tumors from germline deletion carriers.15 Moreover, in three out of four tumors showing biallelic deletions affecting EPCAM the germline and somatic alteration was identical. This strongly supports the hypothesis that gene conversion is a common mechanism of mismatch repair gene inactivation in Lynch syndrome-associated tumors,15 demonstrating that this mechanism of locus-restricted recombination is also responsible for MSH2 inactivation in tumors from EPCAM deletion carriers.
EPCAM MLPA proved applicable for the detection of heterozygous and homozygous deletions in DNA isolated from paraffin-embedded tumor tissue. Our data confirm that MLPA is suitable for germline deletion analysis in DNA isolated from paraffin-embedded tissue,15, 16 thus enabling molecular diagnostics in families affected by Lynch syndrome or other inherited disease conditions, if no blood is available for analysis.
In summary, we demonstrate that lack of EPCAM protein expression in tumors from EPCAM deletion carriers requires biallelic EPCAM deletion, resulting from the combination of a germline and a second somatic deletion, both affecting the EPCAM gene. Lack of EPCAM expression may already occur in precancerous lesions from EPCAM deletion carriers. Our study provides novel information about EPCAM immunohistochemistry in diagnostics of Lynch syndrome and elucidates the mechanism underlying alterations of EPCAM protein expression in EPCAM deletion carriers.
References
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26:5783–5788.
Peltomaki P, Vasen H . Mutations associated with HNPCC predisposition—update of ICG- HNPCC/INSIGHT mutation database. Dis Markers 2004;20:269–276.
Papadopoulos N, Nicolaides NC, Wei YF, et al. Mutation of a mutL homolog in hereditary colon cancer. Science 1994;263:1625–1629.
Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993;75:1215–1225.
Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 1997;17:271–272.
Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994;371:75–80.
Hemminki A, Peltomäki P, Mecklin JP, et al. Loss of the wild type MLH-1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat Genet 1994;8:405–410.
Halvarsson B, Lindblom A, Rambech E, et al. Microsatellite instability analysis and/or immunostaining for the diagnosis of hereditary nonpolyposis colorectal cancer? Virchows Arch 2004;444:135–141.
Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet 2009;41:112–117.
Rumilla K, Schowalter KV, Lindor NM, et al. Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated lynch syndrome cases. J Mol Diagn 2011;13:93–99.
Kloor M, Voigt AY, Schackert HK, et al. Analysis of EPCAM protein expression in diagnostics of Lynch syndrome. J Clin Oncol 2011;29:223–227.
Taylor CF, Charlton RS, Burn J, et al. Genomic deletions in MSH2 or MLH1 are a frequent cause of hereditary non-polyposis colorectal cancer: identification of novel and recurrent deletions by MLPA. Hum Mutat 2003;22:428–433.
Kloor M, Sutter C, Wentzensen N, et al. A large MSH2 Alu insertion mutation causes HNPCC in a German kindred. Hum Genet 2004;115:432–438.
Froggatt NJ, Brassett C, Koch DJ, et al. Mutation screening of MSH2 and MLH1 mRNA in hereditary non-polyposis colon cancer syndrome. J Med Genet 1996;33:726–730.
Zhang J, Lindroos A, Ollila S, et al. Gene conversion is a frequent mechansim of inactivation of the wild-type allele in cancers from MLH1/MSH2 deletion carriers. Cancer Res 2006;66:659–664.
van Dijk MC, Rombout PD, Boots-Sprenger SH, et al. Multiplex ligation-dependent probe amplification for the detection of chromosomal gains and losses in formalin-fixed tissue. Diagn Mol Pathol 2005;14:9–16.
Acknowledgements
The excellent technical assistance of Beate Kuchenbuch, Stefanie Kellner, Petra Höfler, and Daniel Baumgärtner is gratefully acknowledged. This work was funded in part by Grants of the Deutsche Krebshilfe and the Deutsche Forschungsgemeinschaft (DFG, Grant number BL-554/3).
Author contributions
Conception and design: CH, MK, HB; financial support: PS, MvKD, HB; administrative support: PS, MvKD; provision of study materials or patients: MK, CE, HG, MT, PS, MvKD, HB; collection and assembly of data: CH, MK, AV, CE, HG, MT, GB, HB; data analysis and interpretation: CH, MK, AV, HB; manuscript writing: CH, MK, AV, GB, PS, MvKD, HB; final approval of the manuscript: CH, MK, AV, CE, HG, MT, GB, PS, MvKD, HB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Huth, C., Kloor, M., Voigt, A. et al. The molecular basis of EPCAM expression loss in Lynch syndrome-associated tumors. Mod Pathol 25, 911–916 (2012). https://doi.org/10.1038/modpathol.2012.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.30
Keywords
This article is cited by
-
Incidence of germline variants in Lynch syndrome-related genes among Japanese endometrial cancer patients aged 40 years or younger
International Journal of Clinical Oncology (2021)
-
Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy
Journal of Hematology & Oncology (2019)
-
Erblicher Darmkrebs bei Lynch‑/HNPCC-Syndrom in Deutschland
Der Pathologe (2019)
-
Germline deletions in the EPCAM gene as a cause of Lynch syndrome – literature review
Hereditary Cancer in Clinical Practice (2013)
-
Lynch syndrome-associated neoplasms: a discussion on histopathology and immunohistochemistry
Familial Cancer (2013)
