
Abstract
Immunohistochemical staining for DNA mismatch repair proteins may be affected by various biological and technical factors. Staining variations that could potentially lead to erroneous interpretations have been recognized. A recently recognized staining variation is the significant reduction of staining for MSH6 in some colorectal carcinomas. The frequency and specific characteristics of this aberrant MSH6 staining pattern, however, have not been well analyzed. In this study of 420 colorectal carcinoma samples obtained from patients fulfilling the Revised Bethesda Guidelines, we detected 9 tumors (2%) showing extremely limited staining for MSH6 with positive staining present in <5% of the tumor cells. Our analyses showed that these tumors belonged to two distinct categories: (1) MLH1 and/or PMS2 protein-deficient carcinomas (n=5, including 1 with a pathogenic mutation in PMS2); and (2) MLH1, PMS2 and MSH2 normal but with chemotherapy or chemoradiation therapy before surgery (n=4). To test our hypothesis that somatic mutation in the coding region microsatellite of the MSH6 gene might be a potential underlying mechanism for such limited MSH6 staining, we evaluated frameshift mutation in a (C)8 tract in exon 5 of the MSH6 gene in seven tumors that had sufficient DNA for analysis, and detected mutation in four; all four tumors belonged to the MLH1/PMS2-deficient group. In conclusion, our data outline the main scenarios where significant reduction of MSH6 staining is more likely to occur in colorectal carcinoma, and suggest that somatic mutations of the coding region microsatellites of the MSH6 gene is an underlying mechanism for this staining phenomenon in MLH1/PMS2-deficient carcinomas.
Similar content being viewed by others

Main
In recent years, immunohistochemical staining for DNA mismatch repair proteins, MLH1, MSH2, MSH6 and PMS2, has emerged as a simple and useful technique for screening colorectal cancer patients for Lynch syndrome. Using all four antibodies, immunohistochemistry predicts germline mutation with an overall sensitivity of 94%, which is virtually identical to that of PCR-based testing for microsatellite instability.1, 2 It has been recognized that although immunohistochemistry may not detect the mismatch repair deficiency that is not caused by the proteins covered by the four antibodies, it can detect certain mismatch repair-deficient cases that do not exhibit the microsatellite instability phenotype.1, 3
Mismatch repair protein immunohistochemistry, however, may be affected by various biological and technical factors, and the interpretation of these stains may not always be straightforward.1 Yet the immunohistochemical staining results carry significant clinical implications. A stain interpreted as ‘lost,’ ‘abnormal’ or ‘negative’ suggests the potential for a hereditary cancer syndrome and may lead to a costly genetic work-up as well as potentially detrimental psychological impact on the patient. Thus, it becomes imperative that the pathologists be aware of the relevant biological and technical vagaries to achieve the most optimal interpretation of every stain.
Anecdotally, we have observed a patchy, very reduced staining pattern for MSH6 in colorectal carcinomas, particularly in rectal tumors that have undergone neoadjuvant therapy or tumors with mismatch repair deficiency due to loss of other mismatch repair proteins. The proportion of the tumor cells stained may be less than 5%. In one treated tumor case, such a near-complete loss of staining for MSH6 had led to the interpretation of ‘staining absent’; however, testing of the patient’s pre-treatment biopsy sample showed entirely intact staining for the protein in the tumor (unpublished data). As such, this pattern constitutes an interpretation pitfall and deserves the pathologists’ attention.
From a biological point of view, properly interpreting the underlying cause of this phenomenon may allow better understanding of tumor behavior. As the phenomenon happens not just in neoadjuvantly treated tumors but also in tumors that are mismatch repair-deficient due to loss of other mismatch repair proteins, and the MSH6 gene contains a microsatellite of eight mononucleotide repeats in its coding region,4 it seems plausible that a somatic mutation in the coding region microsatellites of the MSH6 gene may be a potential mechanism underlying the marked reduction of protein expression in these cases.
With the goal of better understanding this focal staining pattern of MSH6, we analyzed a consecutive series of colorectal carcinoma samples prospectively accrued from patients fulfilling the Revised Bethesda Guidelines for Lynch Syndrome, and evaluated the frequency and clinical and pathological characteristics of cases exhibiting this focal staining pattern. Furthermore, to explore the hypothesis of somatic mutation being an underlying mechanism, we analyzed tumors demonstrating this staining pattern for the presence or absence of mutation in a (C)8 tract in exon 5 of the MSH6 gene.
Materials and methods
Patient Information
Primary colorectal carcinoma samples were prospectively collected between June 2006 and July 2009 from patients who met the Revised Bethesda guidelines for Lynch Syndrome.5 Specifically, the patients fulfilled at least one of the following three criteria: (1) age equal to or less than 50 years; (2) age between 50 and 60 years, and the colorectal carcinoma showed morphological features suggestive of abnormal DNA mismatch repair as defined previously; and (3) tests requested by the clinician (‘clinical request’) because of a clinical suspicion of Lynch syndrome (ie, patient with synchronous, metachronous colorectal or other Lynch syndrome-associated tumors; with one or more first-degree relatives with a Lynch syndrome-related tumor with one of the cancers being diagnosed <50 years; or two or more first- or second-degree relatives with Lynch syndrome-related tumors regardless of age). In accordance to an institutional protocol, these patient’s colorectal cancer resection specimens were tested for MLH1, MSH2, MSH6 and PMS2 by immunohistochemistry (method described below) at the time of primary tumor resection. These stains were analyzed retrospectively for the purpose of this study. Patients’ clinical information was retrieved from the hospital information system. The study was approved by the Institutional Review Board.
Immunohistochemistry
Immunohistochemistry was performed using the standard streptavidin–biotin–peroxidase procedure. Primary monoclonal antibodies against MLH1 (clone G168-728, diluted 1:250, PharMingen, San Diego, CA, USA), MSH2 (clone FE11, diluted 1:50, Oncogene Research Products, Cambridge, MA, USA), MSH6 (clone GRBP.P1/2.D4, diluted 1:200; Serotec, Raleigh, NC, USA) and PMS2 (clone A16-4, diluted 1:200, BD PharMingen, San Diego, CA, USA) were applied to 5-μm thick 10% formalin-fixed, paraffin-embedded tissue sections. The sections underwent a process of deparaffinization, rehydration and washing in xylene, graded alcohols and distilled water. Blockage of endogenous peroxide activity was performed by incubation with 3% H202. The sections were placed in 10 mM citrate buffer at pH 6 with subsequent microwave antigen retrieval. The antigen–antibody reaction was visualized using the avidin–biotin peroxidase complex (LSAB kit, Dako) and diaminobenzidine as the chromogen. Slides were counterstained with hematoxylin. Non-neoplastic colonic mucosa and colorectal tumors known to be deficient of MLH1, MSH2, MSH6 and PMS2 were used as external positive and negative controls, respectively.
All stains were systematically reviewed for this study. A stain was defined as ‘absent,’ indicating negative protein expression, when there was complete absence of nuclear staining within the tumor cells in the presence of positive staining in the internal non-neoplastic cells. Conversely, positive nuclear staining in the tumor cells was defined as staining ‘present’. MSH6 stains that were classified as staining ‘present’ were further analyzed to identify cases with ‘focal staining,’ defined as mostly negative staining with unequivocal staining present in <10% of the tumor. Cases defined as having the ‘focal staining’ for MSH6 were reviewed for tumor morphology on hematoxylin and eosin (H&E) slides, tumor stage and clinical outcome.
MSH6 Mutation Analysis
The coding region (C)8 microsatellite tract in exon 5 of MSH6 was analyzed in cases with the scanty staining pattern of MSH6 that had sufficient tissue. For each case, a 5-μm section of formalin-fixed, paraffin-embedded tumor tissue was stained by H&E to identify areas of tumor on the slide. Dissection of the tumor area was carried out from the corresponding unstained sections. The tumor tissue was deparaffinized. DNA extraction was performed using the Qiagen DNeasy Tissue Kit (Catalog number 69504, Qiagen). Tumor DNA was amplified using PCR (forward primer: 5′-GAAGACCTATAAAACACTTAGGCTGA-3′; reverse primer: 5′-CCCCCATATTTGGTCCRGTA-3′; R: G and A). PCR conditions consisted of initial denaturing at 95 °C for 10 min; 45 cycles of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s, and a final extension at 72 °C for 7 min. The PCR products were separated by agarose gel electrophoresis to confirm successful amplification. The PCR products were subjected to direct DNA sequence analysis performed by a 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
To analyze the mutant allele frequency in the (C)8 tract in MSH6, the PCR products from tumors showing a (C)8 tract frameshift by direct sequencing was cloned into pCR4 TOPO vectors (Invitrogen, Carlsbad, CA, USA), following procedures of the pCR4 TOPO TA Cloning Kit (Invitrogen). Briefly, the ligation reaction using the TOPO Cloning system contained 1 μl of fresh PCR product, 1 μl of salt solution, 1 μl of TOPO vector and 3 μl of water. The reaction mixture was mixed gently and incubated for at least 5 min at room temperature (longer incubation for up to 1 h would improve the ligation efficiency). One microliter of the ligation reaction was then transformed into DH-5α-competent E. coli provided by the kit. The reaction was kept on ice for 30 min, then heat-shocked for 30 s at 42 °C. The tube was transferred to ice immediately after the heat shock treatment. Two hundred and fifty microliter of room temperature SOC medium was added and the competent cells were plated. The plates were incubated at 37 °C over night. DNA from colonies was amplified using the same primers and PCR condition as DNA from tumor tissue (see above). The PCR products were separated by agarose gel electrophoresis to confirm successful amplification. The PCR products were then subjected to direct DNA sequence analysis performed by a 3730 DNA Analyzer using the forward PCR primer (Applied Biosystems). The number of colonies containing a different number of Cs in the (C)8 tract was counted and recorded.
Results
A total of 420 primary colorectal carcinomas were collected. The patients were composed of 209 males and 211 females with an age range of 18 to 89 years, median age of 47 years and mean age of 48 years. Ninety-eight cancers were proximal to the splenic flexure, 135 from the left colon including 34 from recto-sigmoid, and 187 from rectum.
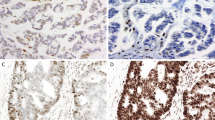
Evaluation of the immunohistochemical stains of the 420 cases showed the following: 92 tumors (92/420, 22%) with ‘staining absent’ for MLH1 and/or PMS2 (66 had concurrent loss of MLH1 and PMS2, 26 lost PMS2 alone), 32 tumors (32/420, 8%) with ‘staining absent’ for MSH2 and/or MSH6 (24 had concurrent loss of MSH2 and MSH6, 8 lost MSH6 alone) and 296 tumors (296/420, 70%) with ‘staining present’ for all four proteins. Further analysis of the MSH6 stains identified 9 tumors (9/420, 2%) with a scanty pattern of staining (‘focal staining’). As detailed in Table 1 and illustrated in Figures 1 and 2, the positively stained tumor cells in all nine cases constituted <5% of the tumor. The positive staining, at least of modest intensity, was often limited to short segments of an individual tumor gland (in gland-forming tumors), or confined to limited foci in tumors with a solid growth pattern. The negatively stained portions were all associated with convincingly positive internal control staining.

Immunohistochemical staining showing scanty MSH6 staining in a colonic adenocarcinoma; a and b represent two areas from one tumor where there is distinct nuclear staining for MSH6, but the staining is present only in a limited number of tumor cells. Note the presence of tumor-infiltrating lymphocytes that stain positively for MSH6. This tumor has intact expression of MSH2 and complete loss of MLH1 and PMS2 (staining not shown).

Immunohistochemical staining for DNA mismatch repair proteins in a rectal adenocarcinoma that has undergone neoadjuvant treatment. (a) Illustration of an area of residual carcinoma present as widely spaced tumor glands in the muscularis propria. By immunohistochemistry, these glands show focal positive staining for MSH6 (b); elsewhere the tumor is negative for MSH6 (staining not shown). This tumor has intact expression of MSH2 (c), MLH1 (d) and PMS2 (staining not shown).
As summarized in Table 1, the clinical and pathological features of these nine cases separated them into two distinct categories: (1) mismatch repair protein deficient and (2) mismatch repair protein proficient but treated pre-operatively.
Cases 1–5 had mismatch repair protein abnormality by immunohistochemistry: three lost MLH1 and PMS2, and two lost PMS2 alone. All five had ‘staining present’ for MSH2. These five cases thus constituted the mismatch repair protein-deficient group. They represented 5% (5/92) of all MLH1/PMS2-abnormal cases. There were one rectal and four colonic carcinomas; none received pre-operative neoadjuvant treatment. The one rectal tumor was judged to be early stage by pre-operative evaluation and, as such, did not undergo neoadjuvant chemoradiation. Upon resection, this tumor was staged as pT2N0. One case (case 4) was tested for germline mutation and was found to carry a PMS2 c.943 C>T (p. R315X) mutation, which was determined to be deleterious. Morphologically, all five tumors showed features characteristic of mismatch repair-deficient colorectal carcinomas. Four tumors exhibited tumor heterogeneity, with the dominant tumor type being medullary in two cases, and conventional (gland forming) and mucinous in one case each. The 5th tumor was a well-differentiated gland-forming tumor. All five tumors had increased tumor-infiltrating lymphocytes (TILs) with a count ≥10 TILs/10 HPFs.
Cases 6–9 had ‘staining present’ for all mismatch repair proteins. All four of them received pre-operative chemotherapy or chemoradiation therapy. Case 6 was a sigmoid colon cancer case that presented with liver metastasis and, as such, the patient received systemic chemotherapy before synchronous primary and metastasis resection. Cases 7–9 were locally advanced rectal cancers that received neoadjuvant chemoradiation before surgery. The pathological treatment response of the four tumors ranged from modest (40%) to profound (90%). All four tumors were of conventional histology with treatment-related changes. None showed apparent TILs.
Adequate DNA samples were obtained from seven of the nine cases with scanty staining for MSH6; two of the four treated tumors failed to yield sufficient DNA.
Analysis of the coding region microsatellite (C)8 in exon 5 of MSH6 in the seven cases identified frameshift mutations in four cases, and all four were mismatch repair-deficient cases (Cases 1–4); the remaining three showed only wild-type alleles (Figure 3). The distribution of the mutant alleles in the (C)8 tract in the four positive cases was further analyzed by cloning and sequencing analysis, and the results are outlined in Table 1. Notably, in each case, the most common allele is the wild-type allele. For the mutant alleles, deletion mutations (−1wt, 9/19) and insertion mutations (wt+1 and wt+2, 10/19) were observed at similar frequencies. One wt+2 allele was observed in case 2. Representative electropherograms of wt, −1wt, wt+1 and wt+2 clones are shown in Figure 4.

Electropherograms depicting the coding region microsatellite (C)8 in exon 5 of MSH6. Illustrations of (a) an intact (C)8 region and (b) an abnormal (C)8.

Electropherograms of the cloned PCR products from tumors with (C)8 frameshift showing the wild-type sequence (a); deletion of 1 C, −1wt (b); insertion of 1 C, wt+1 (c); and insertion of 2Cs, wt+2 (d).
Discussion
The propensity of MSH6 immunohistochemistry to show variable staining patterns is known. In particular, two recent studies have reported specific unusual staining patterns of this protein. Radu et al6 analyzed their challenging cases encountered in colorectal cancer screening for Lynch syndrome and observed a pattern of ‘near-complete loss of MSH6’ in microsatellite instability high tumors and a nucleolar pattern of MSH6 staining in both microsatellite instability high and treated tumors; data about incidence were not provided. A study by Bao et al7 found extensive loss of MSH6 expression in 9 of 51 (18%) treated carcinomas; 6 of the 9 lost staining in 70–80% of the tumor and 3 lost staining in 90% of the tumor. The pre-treatment biopsy samples of all nine tumors showed positive MSH6 staining in >80% of the tumor.
Our study was prompted by our anecdotal experience of cases showing extremely patchy MSH6 staining, leading to erroneous interpretation of ‘staining lost’. Our results extend the observations of Radu et al6 and Bao et al.7 Specifically, we showed that the frequency of scanty staining of MSH6 was about 2% in colorectal carcinoma cases fulfilling the Revised Bethesda Guidelines. In the series we studied, cases exhibiting this staining pattern fell into two distinct categories: mismatch repair protein-deficient tumors and tumors that were mismatch repair-proficient but treated with pre-operative chemotherapy or chemoradiation therapy. Furthermore, our genetic analysis of the cases that showed scanty MSH6 staining detected somatic frameshift mutations in the (C)8 tract of the coding region of the MSH6 gene in mismatch repair protein-deficient tumors but not in mismatch repair-proficient neoadjuvant-treated tumors.
The scanty staining pattern of MSH6 described in this study refers to the extremely limited amount of tumor (<5%) showing positive staining. The positively stained cells may be a single gland, individual cells or a segment of a gland, or patches of cells in tumors with a more solid pattern of growth. As such, the vast majority of the tumor shows absence of any staining for MSH6, indicating a substantial downregulation of protein expression.
Our study has some limitations. The 2% frequency of the phenomenon of scanty MSH6 staining was observed among patients who fulfilled the Revised Bethesda Guidelines, not the general population with colorectal cancer. Furthermore, even for the population meeting the Revised Bethesda Guidelines, the frequency may be somewhat skewed, as the cases ascertained per clinical request did not necessarily follow the stringent Bethesda criteria uniformly. Tumors resected elsewhere were only selectively included (at the discretion of the clinician), as the indication to evaluate these cases was not standardized among the different clinicians. Nonetheless, the 30% (124/420) frequency of mismatch repair protein abnormality identified in our series is consistent with that reported for patient populations meeting the Revised Bethesda Guidelines in the literature,8 suggesting that our patient population is likely to be representative, and the scanty staining pattern of MSH6 is likely an infrequent phenomenon.
As infrequent as this phenomenon may be, awareness of its existence is of great importance in the accurate interpretation of immunohistochemical staining results. As immunohistochemistry is being increasingly used to screen colorectal cancers for the identification of Lynch syndrome, accurate interpretation bears significant clinical implication. It is important for pathologists to be wary about this scanty staining pattern of MSH6 and the clinical scenarios where this pattern is prone to occur. Evidence from our study and others’ suggests that in most clinical scenarios, the scanty staining, that is, marked downregulation of MSH6, is a somatic event and does not suggest germline mutation; the pathologists should not mistake this pattern for the complete loss of MSH6 in the tumor. This will avoid unnecessary germline mutation testing of the MSH6 gene, and consequently avoid the associated psychological burden on the patient.
Notably, however, our results should not be construed as evidence that scanty staining of MSH6 will never be associated with a germline mutation in MSH6. Rather, our data only suggest that when this staining pattern is encountered in colorectal carcinoma, the more likely scenario is mismatch repair deficiency due to abnormal MLH1/PMS2 or effects caused by pre-operative treatment. In the first scenario, the genetic work-up should be first directed to testing for abnormality in the MLH1 or PMS2 genes. In the second scenario, should there be any clinical suspicion of Lynch syndrome, the MSH6 immunohistochemistry should be repeated on the pre-treatment biopsy of the tumor. In both scenarios, genetic work-up of the MSH6 gene may be pursued once these initial attempts have failed to explain the abnormality.
In an attempt to better understand the mechanism underlying the scanty staining pattern of MSH6 in colorectal carcinoma, we analyzed the MSH6 gene for frameshift alterations in its coding region microsatellite sequences and provided evidence associating the scanty staining with somatic frameshift mutations in the (C)8 tracts of the coding region of MSH6 in mismatch repair-deficient colorectal cancers, but not in mismatch repair-proficient and previously treated tumors. This is consistent with our current knowledge about the MSH6 gene and the function of DNA mismatch repair system.9
It is known that the MSH6 is one of approximately 32 target genes in the human genome that contain a mononucleotide repeat of seven or more elements in the coding sequence.4 Such coding region microsatellites have been shown to have the propensity for undergoing spontaneous frameshift mutations during DNA replication,9, 10 and the replication fidelity of these sequences is dependent upon the DNA mismatch repair system.11 Previous studies have demonstrated that microsatellite-unstable gastrointestinal cancers have a higher incidence of somatic frameshift mutations in the MSH6 gene. It is plausible, therefore, that in our cases with defective MLH1 and/or PMS2, the reduced MSH6 protein expression as reflected by the scanty staining pattern is a manifestation of the somatic frameshift mutation in the (C)8 tract of the coding region of MSH6.
The biological implication of somatic MSH6 mutations in mismatch repair-deficient colorectal carcinoma is yet to be determined. In this study, all tumors that had scanty MSH6 staining, that is, marked reduction of MSH6 protein, showed characteristic morphological features of microsatellite instability high cancers. Clinically, the tumors presented as stage II or III cancers with variable clinical outcome. The limited number of cases precludes any meaningful conclusions and determination of the clinical significance awaits additional observations.
Secondary mutation in the (C)8 tract of the MSH6 gene, however, is not likely to be the reason for the scanty staining pattern of MSH6 in mismatch repair-proficient, previously treated colorectal carcinomas. In such cases, two alternative explanations may be considered. First, it may be possible that somatic mutations elsewhere in the MSH6 gene occur as a consequence of treatment effect. In this regard, it is intriguing to note that The Cancer Genome Atlas indeed identified post-treatment MSH6 mutations in glioblastomas, and studies thus far have suggested that inactivating MSH6 mutations in glioblastomas treated with temozolomide are selected during therapy and are causally associated with temozolomide resistance.12, 13 Second, the downregulation of MSH6 gene may be related to tissue hypoxia in treated tumors. Given that hypoxia and oxidative stress have been demonstrated to impair mismatch repair function in genetically mismatch repair-proficient tissues,14, 15, 16, 17 it is possible that hypoxia in a tumor subjected to the cytotoxic effects of chemotherapy or chemoradiation therapy could lead to downregulation of the MSH6 protein. Research efforts to explore these mechanisms are underway in our laboratory. It is to be anticipated that as the understanding of the biological and biochemical properties of the DNA mismatch repair proteins progresses, and as biotechnology continues to advance, better methods in detecting and managing Lynch syndrome patients will be achieved.
References
Shia J . Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn 2008;10:293–300.
Boland CR, Shike M . Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology 2010;138:e1–e7.
Zhang L . Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J Mol Diagn 2008;10:301–307.
Boland CR, Koi M, Chang DK, et al. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Fam Cancer 2008;7:41–52.
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261–268.
Radu OM, Nikiforova MN, Farkas LM, et al. Challenging cases encountered in colorectal cancer screening for Lynch syndrome reveal novel findings: nucleolar MSH6 staining and impact of prior chemoradiation therapy. Hum Pathol 2011;42:1247–1258.
Bao F, Panarelli NC, Rennert H, et al. Neoadjuvant therapy induces loss of MSH6 expression in colorectal carcinoma. Am J Surg Pathol 2010;34:1798–1804.
Pinol V, Castells A, Andreu M, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. J Am Med Assoc 2005;293:1986–1994.
Malkhosyan S, Rampino N, Yamamoto H, et al. Frameshift mutator mutations. Nature 1996;382:499–500.
Gifford G, Paul J, Vasey PA, et al. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res 2004;10:4420–4426.
Ohmiya N, Matsumoto S, Yamamoto H, et al. Germline and somatic mutations in hMSH6 and hMSH3 in gastrointestinal cancers of the microsatellite mutator phenotype. Gene 2001;272:301–313.
Hunter C, Smith R, Cahill DP, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006;66:3987–3991.
Yip S, Miao J, Cahill DP, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 2009;15:4622–4629.
Chang CL, Marra G, Chauhan DP, et al. Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol Cell Physiol 2002;283:C148–C154.
Bindra RS, Crosby ME, Glazer PM . Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev 2007;26:249–260.
Rodriguez-Jimenez FJ, Moreno-Manzano V, Lucas-Dominguez R, et al. Hypoxia causes downregulation of mismatch repair system and genomic instability in stem cells. Stem Cells 2008;26:2052–2062.
Mihaylova VT, Bindra RS, Yuan J, et al. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol 2003;23:3265–3273.
Acknowledgements
This study was supported in part by The Colon Cancer Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Presented in part at the annual meeting of the United States and Canadian Academy of Pathology, Washington, DC, USA, 20–26 March 2010, and the annual meeting of the Association for Molecular Pathology, Grapevine, TX, USA, 17–19 November 2011.
Rights and permissions
About this article
Cite this article
Shia, J., Zhang, L., Shike, M. et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1/PMS2 deficiency. Mod Pathol 26, 131–138 (2013). https://doi.org/10.1038/modpathol.2012.138
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.138
Keywords
This article is cited by
-
An unusual phenotype occurs in 15% of mismatch repair-deficient tumors and is associated with non-colorectal cancers and genetic syndromes
Modern Pathology (2022)
-
Somatic hits in mismatch repair genes in colorectal cancer among non-seminoma testicular cancer survivors
British Journal of Cancer (2022)
-
Detecting mismatch repair deficiency in solid neoplasms: immunohistochemistry, microsatellite instability, or both?
Modern Pathology (2022)
-
Concordance analysis of microsatellite instability status between polymerase chain reaction based testing and next generation sequencing for solid tumors
Scientific Reports (2021)
-
Identification of Lynch syndrome-associated DNA mismatch repair-deficient bladder cancer in a Japanese hospital-based population
International Journal of Clinical Oncology (2021)
