
Abstract
Mucosal immune responses must be tightly controlled, particularly in the intestine. As members of the mononuclear phagocyte family, dendritic cells (DCs) and macrophages are well represented in intestinal tissues and have developed unique functional niches. This review will focus on recent findings on antigen uptake and processing in the intestine and the role of DCs in the imprinting of homing receptors on T and B cells, the induction of immunoglobulin A B-cell responses, and the differentiation of regulatory T cells. It will also address the unique phenotype of intestinal macrophages and briefly what is known regarding the relationships between these cell types.
Similar content being viewed by others
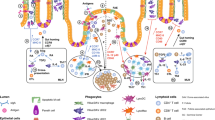

Introduction
In mucosal tissues, the development of inflammation and immunity is central to effective host defense against invading pathogens, yet must be tightly regulated to prevent abnormal responses to innocuous environmental antigens and commensal organisms that result in allergy or chronic inflammatory diseases. This is particularly true in the intestine, where there is continuous exposure to a vastly complex mixture of ingested environmental and food antigens, and intestinal microbiota.
This review will highlight recent findings regarding the phenotype and function of dendritic cells (DCs) and macrophages in the induction and regulation of immune responses in the intestine. DCs likely have a key role in immune regulation as they are prominently localized to mucosal surfaces, both at sites of antigen uptake and within inductive lymphoid tissues, and have been shown to process antigens given in both tolerogenic and immunogenic forms, as well as to directly sample endogenous flora and pathogenic microorganisms in vivo. Furthermore, sub-populations of mucosal DCs have unique functions when compared to DCs from non-mucosal sites. These include the imprinting of mucosal homing receptors on T and B cells, the induction of regulatory T cells (Tregs) to soluble antigens in the resting or “steady” state, and the direct contribution to immunoglobulin A (IgA) B cell class switching. Mucosal macrophages are also found prominently in the intestine, primarily within the lamina propria (LP), and are particularly capable of taking up and killing bacteria. Evidence also indicates that intestinal macrophages have immunoregulatory roles, including the production of suppressive cytokines that affect DC function, and the potential to directly induce the differentiation of Tregs.
Antigen Uptake and Cell Trafficking
Important for understanding how mucosal immune responses are induced and regulated is the issue of where different types of antigens are taken up and presented to T and B cells. Primary sites for the induction of intestinal T and B cell responses are Peyer's patches (PPs) in the small intestine, isolated lymphoid follicles (ILFs) in the small and large intestine, and mesenteric lymph nodes (MLNs). In contrast, the diffuse LP and the intraepithelial cell compartments are primarily effector sites. One caveat to this conclusion, however is that recent evidence indicates that B cells may undergo class-switch recombination directly in the LP (see below).
Luminal antigens, including macromolecules, bacteria, and viruses gain access to the cells of PPs and ILFs through specialized antigen-transporting epithelial cells, M (micro-fold) cells, present in the follicle-associated epithelium (FAE) above organized lymphoid structures of most mucosal tissues.1, 2 M-cell transport is promiscuous and mediated by binding to surface-expressed carbohydrates in regions free of overlying mucus, but can be enhanced by the presence of antigen-specific IgA,3, 4, 5 by immune targeting with anti-M-cell antibodies,6 or by oral administration of toll-like receptor (TLR) 2 or TLR4 ligands.7, 8
Dendritic cells are present within the FAE in small numbers and in large numbers in the subepithelial dome (SED).9 Furthermore, orally administered cholera toxin, cholera toxin B-subunit, or Escherichia coli heat-labile toxin,10 and proteosome vaccines8 as well as Salmonella typhimurium infection11 can induce an influx of DCs from the SED into the FAE. In the latter study, PP DCs in the SED that express CCR6 appear to migrate into the FAE, where they form clusters with antigen-specific CD4 T cells. Furthermore the activation and expansion of specific T cells was dependent on CCR6.11 In contrast, CCR6-expressing DCs were not recruited to the intestinal LP, suggesting that CCD6+ DCs may have specific functions in organized lymphoid structures, where the CCR6 ligand, CCL20, is constitutively expressed.12
Peyer's patch DCs in the SED capture soluble antigens given orally9, 13 and take up, or are initial targets of, orally administered pathogens, including S. typhimurium,14, 15 Listeria monocytogenes,16 Brucella abortus,17 and Helicobacter pylori.18 Furthermore, PP DCs in the SED take up apoptotic epithelial cells following intestinal reovirus infection.19
Following activation, DCs in the SED can migrate to T-cell interfollicular regions (IFR), as shown following oral CT or systemic administration of a soluble antigen preparation from Toxoplasma gondii tachyzoites (STAg).12 In the later study, PP DCs downregulated CCR6, expressed CCR7, and migrated to the IFR where CCR7 ligands, CCL19 and CCL21, are expressed.12
Increasing data also suggest that PPs are primary sites for the uptake of commensal bacteria and that DCs may be primary targets. Following oral administration of Enterobacter cloacae, organisms were found in DCs in the PPs and MLNs, but not in the LP or spleen, where they likely help drive IgA production that then overall limits their contact with epithelial cells and penetration into the LP.20 Organisms were not found in presumed macrophages (CD11b+ CD11c− cells) from these tissues, and in contrast to DCs, ex vivo CD11b+CD11c− cells were able to kill E. cloacae efficiently. This indicated that commensal bacteria can be taken up and persist in DCs in PPs (and possibly ILFs) and MLNs, but cannot migrate to systemic sites, thus limiting their potential to cause systemic inflammation.20 Furthermore, specific IgA induced against commensal bacteria may target these organisms for limited, directed uptake into PPs and ILFs.5 Interestingly, initial erosions in ileal Crohn's disease appear to occur over lymphoid follicles, and uptake of nonpathogenic E. coli by FAE is enhanced in patients with long-standing disease, suggesting that abnormal uptake and/or poorly regulated immune responses to commensal bacteria in PPs may be involved in early pathogenesis of Crohn's disease.21, 22
The extent to which PP DCs traffic to the MLNs is not yet clear; however, the presence of chemokines, such as CCL19 and CCL21, important for DC migration to LN within the T-cell zones of PPs12 suggests that PP DC migration, as well as specific immune responses, may be relatively contained within the PP. Furthermore, phenotypic analysis of DC sub-populations in the rat indicates that the primary source of migratory DCs is the intestinal LP and not the PPs.23
A second site for antigen entry into the intestine is the nonfollicular absorptive epithelium, where both soluble antigens and bacteria can gain access to DCs in the LP. This can occur by trans- or paracellular transport, by receptor-mediated trafficking, such as occurs through the neonatal FcR expressed on absorptive epithelial cells in humans,24 by direct sampling of luminal contents by DC extensions that reach between epithelial cells into the intestinal lumen25, 26, 27 or by direct damage to the epithelium, as can occur during inflammatory bowel disease, or by infection with human immunodeficiency virus28 or Shigella flexneri.29 Antigen sampling may also occur by uptake of exosomes from epithelial cells or across villous M cells.30
Even in the absence of infection or inflammation, LP DCs constitutively traffic to MLNs,31 which appears to be a relatively active process. These migratory DCs can carry self- or cell-associated antigens from apoptotic epithelial cells32 or soluble proteins given orally.33, 34, 35 Soluble antigens given orally can be processed by LP DCs, which then migrate to the MLN in a CCR7-dependent manner, which was shown to be essential for oral tolerance induction.35 In addition, commensal bacteria are present within LP DCs, and both S. typhimurium and noninvasive E. coli can be taken up by luminal dendrites in the terminal ileum when given orally,25, 26 which may carry these bacteria to the MLN. Interestingly, it was recently shown that the major route of entry into the body of noninvasive S. typhimurium may indeed be through DC extensions, whereas pathogenic S. typhimurium preferentially invade PPs to induce systemic or mucosal humoral immune responses, respectively.36
Activation of LP DCs results in enhanced migration to MLNs, as occurs following systemic administration of lipopolysaccharide or orally administered TLR7/8 agonists.37,38 Furthermore, when compared with DCs from other sites, LP DCs preferentially express TLR5,39 which on activation by bacterial flagellin,40 may be important for DC activation and migration to MLNs or spread of invasive bacteria to systemic sites.39 These studies indicate that LP DCs are fully capable of becoming activated following administration of microbial products, resulting in enhanced migration to MLN. However it is not yet clear whether this effect is direct or mediated by inflammatory cytokines produced by other cells.
The Expanding Family of Intestinal DC Populations and Their Specialized Functions
Multiple DC sub-populations have been identified in the PP, MLN, and intestinal LP, which differ in their surface phenotype, localization, cytokine production, and ability to drive T-cell differentiation in vitro. These studies have been the subject of several recent reviews41, 42, 43, 44 (and see Table 1). Over the past several years, several unique functions of intestinal DCs have been identified including the imprinting of lymphocytes with unique homing receptors that allow for their recirculation to intestinal tissues, the capacity to provide direct signals for the differentiation of IgA-producing B cells, and the ability to drive the differentiation of regulatory T cells that are involved in tolerance to soluble oral antigens and commensal bacteria.
Induction of Homing Receptors
Initial studies implicating the importance of the lymphoid microenvironment in the imprinting of homing receptors on T cells demonstrated that adoptively transferred TCR transgenic CD4+ and CD8+ T cells primed in the mesenteric, but not cutaneous LNs or spleen, expressed high levels of α4β7, an integrin that binds to MadCAM-1 expressed on high-endothelial venules of intestinal tissues, and CCR9 and migrated in response to CCL25, the chemokine ligand of CCR9 expressed constitutively in the small intestine.45, 46, 47 Subsequently, it was shown that T cells primed in vitro with antigen-pulsed MLN or PP DCs, but not DCs from the spleen or peripheral lymph nodes, expressed CCR9 and high levels of α4β7.46, 48, 49 Furthermore, PP DC-primed CD8+ T cells had enhanced migration to the small intestine,48 which was dependent on a fixation-sensitive signal from the DCs.50
The capacity of intestinal DCs to drive intestinal homing receptors on T cells is largely dependent on retinoic acid (RA), a metabolite of retinol (vitamin A). Dietary retinol or retinoids hydrolyzed to retinol are stored in the liver and released at a constant level in the blood. Retinol becomes successively oxidized inside cells to retinal by alcohol dehydrogenases or members of the short-chain dehydrogenase/reductase family and then to RA by retinal dehydrogenases. RA signaling is mediated by nuclear receptors of the RA receptor (RAR) and retinoid X (RXR) families, which form RAR/RXR heterodimers. On RA binding to the RAR, the RAR/RXR heterodimers act as transcription factors.
In initial studies, exogenous RA directly drove the expression of α4β7 and CCR9 on T cells activated in vitro with anti-CD3 and anti-CD28, which homed to the intestine.51 Furthermore, PP and MLN DCs expressed retinal dehydrogenases, produced RA from retinol, and inhibitors of RA production and signaling blocked DC-induced α4β7 and CCR9 expression. Finally, mice on vitamin A-deficient diets had a reduction in α4β7+ memory T cells in lymphoid organs and a dramatic deficiency of LP T cells in the small intestine.51 Interestingly, epithelial cells in PPs also expressed high levels of an isoform of retinal dehydrogenase, RALDH1, indicating that RA production is not likely restricted to DCs. In addition, it is possible that RA from epithelial cells conditions local DCs for the ability to produce transforming growth factor-β (TGF-β) and interleukin (IL)-6 and the capacity to augment mucosal homing receptor expression.52
In studies from two laboratories, intestinal DCs expressing CD103, the αE compenent of the αEβ7 integrin, were shown to have unique functional properties.53, 54 In studies of homing receptor expression, LP DCs were demonstrated to be as potent as MLN DCs in inducing α4β7 on CD8+ T cells and better at inducing CCR9. Extensive phenotypic analysis identified particularly high numbers of DCs expressing CD103 in the LP compared with the MLN.54 Furthermore, CD103+ DCs from the MLN were found to express higher levels of major histocompatibility complex II than the same cells from the LP and to be far lower in number in the MLN of mice lacking CCR7, indicating that this population likely migrates from the LP to the MLN in the steady state. When tested in vitro, only CD103+ DCs from either the MLN or LP were found to drive the expression of high levels of CCR9 and α4β7 on CD8 T cells. Furthermore, CCR9 and α4β7 were not induced on CD8+ T cells in MLNs of CCR7-deficient mice given systemic antigen and lipopolysaccharide, despite equivalent levels of proliferation of the CD8+ T cells in the MLN.
In studies in the T-cell transfer model of colitis, CD103 expression by host cells was essential for the ability of CD4+CD25+ regulatory T cells to protect against colitis induction.53 DCs were the predominant host cells expressing CD103 in the spleen, and high numbers of CD103+ DCs were found in the colon and MLN. Interestingly, CD103 expression was found on the three previously identified DC subsets from the spleen, MLN, and colon, which were CD8+/CD11b−, CD8−/CD11b+, and CD8−/CD11b−, with the highest percentage of CD103+ DCs in the CD8+ population. Functional studies demonstrated that CD103+ DCs, but not their CD103– counterparts, promoted expression of the gut-homing receptor CCR9 on CD4+ T cells. Collectively, these studies indicated that a sub-population of MLN DCs expressing CD103 was likely derived from the intestinal LP and, on migration to MLNs, were essential for driving homing receptors on CD4+ and CD8+ T cells.
Most recently, studies using RA-responsive element reporter mice demonstrated that whereas both spleen and MLN DCs were capable of driving RAR signaling and α4β7 expression in CD8+ T cells, only CD103+ DCs from the MLN drove an early RAR signal that was required for CCR9 and high levels of α4β7 expression in vitro.55 Furthermore, CD8+ T cells primed in vivo in the MLN had evidence of enhanced RAR signaling. Interestingly, DC-mediated induction of gut-homing receptors was inhibited on CD8+ T cells at a high antigen dose without influencing RAR signaling events, indicating that the induction of gut-homing receptors is likely controlled by the intensity of both RAR signaling and antigen dose. These data implied that the early and high levels of RA production by mucosal CD103+ DCs are largely responsible for their ability to drive CCR9 and α4β7 expression on activated lymphocytes, and that this can be overcome by high antigen doses.
Finally, similar to what was shown for T cells, PP and MLN DCs induced CCR9 and α4β7 expression on both naive- and antigen-experienced B cells stimulated with anti-IgM. This was dependent on RA, and allowed B cells to selectively home to the intestine.56 Furthermore, repeated stimulation of human naive B cells with spleen DCs with and without RA demonstrated flexibility in homing receptor expression, in that cells initially stimulated in the absence of RA still expressed high levels of CCR9 and α4β7 on repeated activation in the presence of RA and vice versa. This implicated gut-associated lymphoid tissue DCs in driving homing receptors on both T and B lymphocytes, which was dependent on RA.
Induction of IgA Responses
A central function of the mucosal immune system is the production of IgA. High-affinity IgA acts to exclude microorganisms and toxins from entering the body, whereas low-affinity IgA is thought to inhibit the binding of commensal bacteria to epithelial cells. Furthermore, the latter may be important in the maintenance of an appropriate intestinal microbiota.57
Naive B cells are induced to undergo class switch recombination and somatic hypermutation in organized lymphoid tissues of the intestine; however, recent data indicate class switch recombination may also occur in the diffuse LP, where activation-induced cytodine deaminase (AID), an essential enzyme for isotype switching, has been detected in B cells in some, but not all studies (see ref. 57). In organized tissues, such as the PPs, MLNs, and ILFs, local factors including TGF-β, IL-6, and IL-10, together with activation of CD40 by T-cell-expressed CD40L, which drives AID expression, are thought to be responsible for T-cell-dependent IgA B-cell differentiation. In the LP, TLR-dependent induction of innate IgA class switch recombination-inducing factors, including APRIL (a proliferation inducing ligand) and its homolog B cell-activating factor of the tumor necrosis factor family (BAFF, also known as BLyS) from epithelial cells, DCs, and B cells, is thought to drive T-cell-independent IgA B-cell differentiation.
Recent studies have implicated a unique role for mucosal DCs in regulating IgA B-cell differentiation. First, bacteria are present in lymphoid follicles, including PPs, and in the LP in the terminal ileum of normal mice, and can be found within DCs.20, 58, 59, 60 In addition, both humans and mice produce significant amounts of secretory IgA against commensal bacteria, which is induced in the absence of T cells.20 Second, PP DCs were recently shown to not only induce homing receptors on B cells (as noted above), but also to drive IgA B cell differentiation in the absence of T-cell signals through the production of RA and IL-6.56 Furthermore, DCs from peripheral LNs or liver were capable of inducing IgA B cell differentiation in the presence of exogenous RA and either IL-5 or IL-6, but, interestingly, not TGF-β. Finally, vitamin A-deficient mice had a paucity of IgA+ B cells in the LP, but normal numbers of naive IgM+ B cells in PPs.56 Therefore, mucosal DCs can contribute to direct IgA B cell differentiation to commensal bacteria, either in lymphoid structures or in the LP through their ability to produce RA- and TLR-induced IL-6, and possibly BLyS/APRIL. Furthermore, their ability to produce TGF-β, as well as their superior capacity to activate naive T cells, most certainly indicates a central role for DCs in T-cell-dependent IgA B-cell differentiation in organized lymphoid structures. In support of this possibility, CD11b+ DCs from PPs were shown to be superior to other DC populations in driving IgA B-cell differentiation in the presence of cognate T cells in vitro, a function dependent on their ability to produce IL-6.61
Oral Tolerance and The Peripheral Differentiation of Regulatory T Cells
Several mechanisms have been identified by which systemic tolerance to orally administered antigens (oral tolerance) is induced, which have been largely identified in mice fed soluble proteins, such as ovalbumin, myelin basic protein, retinal S-antigen, collagen, and insulin, as well as peptides.62, 63 These mechanisms include the induction of T-cell anergy, deletion, and the induction of CD4+ Tregs. The mechanisms involved are influenced by antigen dose and frequency with higher doses favoring anergy and deletion, and lower and repeated doses favoring the generation of Tregs capable of transferring to naive mice tolerance to subsequent immunization or the induction of autoimmune disease (see ref. 62). Antigen-specific Tregs capable of bystander suppression were initially identified as Th3 cells producing TGF-β (see ref. 64). Later studies demonstrated that oral antigen administration could also induce the differentiation or expansion of antigen-specific CD4+CD25+ Tregs in the MLN that could transfer tolerance to naive mice.65, 66 Furthermore, naturally occurring CD4+CD25+ T cells were required for oral tolerance in a CD8+ T-cell-dependent model of skin contact hypersensitivity.67
Initial studies implicating DCs in oral tolerance demonstrated that expansion of DCs in vivo with Flt3L administration resulted in an enhanced sensitivity to oral tolerance induction.68 In addition, PP DCs could be loaded with soluble oral antigens,9, 13 and DCs from the PPs, but not the spleen, were shown to produce IL-10 and likely TGF-β,69 and both PP DCs69 and MLN DCs from fed mice70 were able to drive the differentiation of T cells producing IL-4 and IL-10 in vitro. Furthermore, PP CD11b+ DCs were unique in their ability to produce IL-10 and drive the differentiation of IL-10- and IL-4-producing T cells.71 Finally, DCs from the LP were shown to take up oral antigens, to express mRNA for IL-10 and interferon-γ, but not IL-12, and, following antigen feeding, were able to induce tolerance when transferred to naive mice.72 Finally, data indicated that plasmacytoid DCs from the PP were able to drive the induction of IL10-producing T cells in vitro.122 Together, these studies indicated that PP and LP DCs may drive noninflammatory T-cell responses, as well as tolerance following antigen feeding.
Recently, significant new studies demonstrate that mucosal DCs are able to drive de novo induction of CD4+Foxp3+ Tregs.73, 74, 75 CD4+Foxp3+ Tregs can differentiate in the thymus in response to self-antigens expressed at this site during a restricted period of postnatal development.76 In addition, CD4+FoxP3+ Tregs can differentiate from CD4+CD25− T cells in vitro in the presence of TGF-β,77, 78, 79 which are functionally relevant as they can suppress experimental colitis induction.80 Most recently, peripheral induction of Foxp3+ Tregs was found to occur primarily in lymphoid tissues associated with the intestine (PPs, MLNs, and small intestinal LP).73, 81 Importantly, following adoptive transfer of naive TCR transgenic T cells to normal mice, oral antigen feeding resulted in the accumulation of antigen-specific Foxp3+ Tregs in the PP, MLN, and small intestinal LP,81 implicating peripheral Treg induction in oral tolerance to soluble proteins.
Interestingly, DCs from mucosal tissues (MLN or small intestine LP) were more capable than spleen DCs of inducing Foxp3 expression in the presence of exogenous TGF-β,74, 81, 82 and MLN DCs were less capable of inducing Th17 cells in the presence of TGF-β and IL-6.74 Furthermore, CD103+ DCs, but not CD103− DCs, from the small intestinal LP or MLN were shown to induce the differentiation of Tregs in the absence of exogenous cytokines.73, 81 This occurred by their production of RA and TGF-β.73, 81 The induced Foxp3+ Tregs were as efficient as naturally occurring Tregs in suppression assays in vitro and in vivo.
RA also enhanced the in vitro generation of Foxp3+ Tregs from naive T cells in the presence of IL-2 and TGF-β in the absence of DCs.83, 84 Furthermore, the addition TGF-β and RA, but not RA, alone to spleen or CD103− MLN DCs was able to significantly enhance the differentiation of Foxp3+ Tregs, whereas TGF-β alone has modest enhancing effects, indicating that RA likely acts to enhance TGF-β-mediated Treg differentiation.73, 74, 81
In addition to positive effects on Treg differentiation, RA was able to suppress Th17 induction in the presence of IL-6 and TGF-β,74, 83 and IL-6 was capable of inhibiting Foxp3 induction with TGF-β and RA, depending on the doses of RA and IL-6 in the cultures.74 This suggests that IL-6 (and possibly other cytokines, e.g., IL-1, TNF-α) acts in a reciprocal fashion with RA to control Treg induction in the presence of TGF-β-rich environment of the intestine.
In contrast to CD103+ DCs, CD103− DCs induced Th1 differentiation and produced significant amounts of IL-6, TNF-α, and IL-23 in response to lipopolysaccharide or CD40 signaling, whereas CD103+ DCs were much less responsive to stimulation and were shown to contain enzymes (ALDH1A2) involved in the conversion of retinol to RA.73 In contrast, both CD103+ and CD103− DCs in the intestinal LP were capable of driving nearly equivalent Treg differentiation in the presence of TGF-β that was dependent on RA.81 Therefore, consistent with studies of intestinal homing receptor induction, it appears that CD103+ DCs in the MLN may be derived from the intestinal LP, whereas the MLN CD103− DCs may have come directly from blood precursors.73 Furthermore, the LP and not the MLN microenvironment may be important for DC conditioning for Treg induction.
Intestinal Macrophages
Although both DCs and macrophages are members of the mononuclear phagocyte system, DCs can be distinguished from macrophages based on their dendritic morphology, their efficient ability to capture, process and present antigens to naive T cells, and their unique life cycle, acting as sentinels in peripheral tissues that on tissue or microbial signals carry self- and foreign antigens to draining lymphoid tissues for the induction of T-cell tolerance and immunity. As such, DCs have developed unique endocytic system for processing of antigens, are localized in many peripheral sites exposed to the external environment, and go through a defined process of maturation in response to a variety of stimuli. Macrophages belong to a vast family of tissue cells, including Kupffer cells in the liver, and glial cells in the brain, but generally share several general functional attributes, at least in many tissues in which they are found. In particular, they have predominantly innate immune functions, such as the capturing and killing of microbes, the scavenging of apoptotic and dead cells, and the production of regulatory cytokines. They are also less efficient at presenting antigen to T cells. What is clear, however, is that the phenotype and functions of both DCs and macrophages can vary depending on the tissue and the presence of tissue injury, inflammation, and exposure to microorganisms and external antigens. In addition, there is significant overlap in the origins, surface characteristics, and many functional attributes of these cell types, which is becoming more apparent as more sub-populations of these cell types are defined.
Macrophages have been defined in the intestinal tract in both mice and humans, and are present in high numbers. In fact, in early studies, based on the expression levels of the F4/80 glycoprotein, which is present on circulating monocytes as well the vast majority of tissue macrophages in the mouse, the small and large intestines contained by far the largest reservoir of these cells in the body.85 F4/80+ cells are found extensively in the small and large intestine, where they are in close contact with the epithelium, and express CD11b and low-to-modest levels of major histocompatibility complex II.86, 87 F4/80+ cells have also been identified in PPs of mice, however, are present at very low numbers and are found at the follicle base near the draining lacteals.9 Of note is the fact that F4/80 is also expressed on some “DC” populations, such as Langerhans cells, so the extent to which this marker differentiates cell types in the murine intestine is not yet clear. In other species, including humans, highly phagocytic cells described as macrophages, containing bacteria, are also found in the SED of the PP. In humans, LP macrophages in the colon to express CD11b and low levels of major histocompatibility complex II, as well as microsialin (CD68) and low levels of CD11c.88 In the human small intestine, macrophages express high levels of major histocompatibility complex II and CD13, a zinc metalloproteinase, but negligible levels of CD11b and CD11c.89
Intestinal macrophages from humans and mice appear to share several unique characteristics in the steady state. First, they avidly phagocytose particulate antigens and bacteria, and are highly active in killing these organisms.20, 89 Second, they are highly suppressed in their responses to activating signals from cytokines or pathogens, including TLR ligands, that typically induce the production of proinflammatory mediators and cytokines and enhance the antigen-presenting capacity of circulating monocytes (see ref. 90 and ref. 91). This includes the poor proinflammatory cytokine and chemokine production, as well as the lack of inducible expression of costimulatory molecules. Intestinal macro-phages lack or have poor expression of most innate response receptors, notably CD14, Fcγ, and Fcα receptors, TLR1-5 and TREM-1. In addition, they have suppressed nuclear factor-κB signaling, possibly due to the presence of suppressive cytokines, such as IL-10 or TGF-β, similar to what has recently been shown for DC populations.92
Furthermore, mouse intestinal macrophages produce IL-10, both constitutively93 and following stimulation with bacteria.87 In fact, IL-10 may be essential for this poor responsiveness of intestinal macrophages in mice, as intestinal macrophages from IL-10−/− mice have enhanced cytokine production in response to TLR-ligand stimulation.87, 93, 94 Therefore, intestinal macrophages in the steady state are noninflammatory cells that retain the capacity to phagocytose and kill invading microbes.
Recent data in mice suggest that intestinal macrophages may also have the capacity to induce Foxp3+ Tregs.93 Thus, it was shown that intestinal macrophages (CD11cloCD11b+F4/80+ cells) from the murine small intestine produced IL-10, and induced Foxp3+ Tregs from naive T cells in culture, but only significantly in the presence of exogenously added TGF-β. Interestingly, in this study, Treg induction was IL-10 dependent, and DC populations, including CD11b−CD103+ DCs, were not capable of inducing Tregs in the absence of TGF-β. Furthermore, CD11b+ DCs, which were identified to be primarily CD103−, induced Th17 cells, which was suppressed by co-culture with intestinal macrophages. Although it is not yet clear why this study provided discrepant results from studies from other laboratories with regard to CD103+ intestinal DC function,73, 75 it indicates an additional suppressive function of intestinal macrophages. Local macrophages may be capable of supporting Treg and blocking Th17 induction within the local tissues.
During active intestinal inflammation, as occurs in Crohn's disease in humans, blood monocytes appear to be recruited to inflamed tissues where they release a variety of proinflammatory cytokines, such as TNF-α, macrophage infiltrating factor, IL-1, IL-6, IL-12, and IL-18, which are critically involved in the onset and the development of Crohn's disease. Furthermore, during infectious inflammation of the intestine, such as that occurring with S. typhimurium infection in the mouse, a population of CD11cint CD11b+ cells is recruited to PPs that produce TNF-α that may be derived from monocytes.95 These cells may be similar to the cells recruited to the spleen during listeria infection that produced TNF-α and inducible nitric oxide synthase (so-called “Tip DCs”).96 Recently, these same cells have been implicated in the regulation of IgA production in mucosa-associated lymphoid tissues.97
Development of Intestinal DC Populations
Comparatively little is known about the developmental and functional relationships between DC populations and macrophages in the intestine, in particular when compared with what is known about the spleen or skin.
This issue was recently addressed in cell transfer studies aimed at identifying the progenitors of LP DCs. It is now clear that two sub-populations of blood monocytes exist in mice, humans, and rats, one that is CCR2+ CX3CR1int and the other that is CCR2−CX3CRIhi (see ref. 98). Both subsets in the mouse express F4/80 and CD11b. In addition, CCR2+ cells also express high levels of LyC6 (stained with GR1 antibody) and were originally shown to migrate to inflammatory sites (so-called “inflammatory” monocytes), whereas CCR2− cells express low levels of Ly6C and migrate constitutively to noninflammed sites; both have the potential to give rise to DCs.99 In addition, the same group isolated a common progenitor from the bone marrow that has the capacity to differentiate into both DCs and monocytes (monocyte and dendritic cell progenitor, MDP).100 Using adoptive transfer strategies with these defined cell populations,101 it was demonstrated that CCR2+Ly6Chi monocytes given intravenously to normal mice resulted in no detectable spleen or LP DCs derived from the transferred cells. However, when given to mice depleted of CD11c cells using the CD11c diphtheria toxin receptor transgenic mice given diphtheria toxin, LP DCs were readily replenished by transferred monocytes. In contrast, spleen DCs were only repopulated when given MDP and not CCR2+Ly6Chi monocytes. The same was true for mice made genetically deficient for CD11c+ cells.101 Furthermore, all monocyte-derived DCs were CX3CR1+ cells, similar to what has been shown for a large portion of LP DCs from normal mice, and in particular those that extend dendrites into the intestinal lumen.26
These studies indicate that monocytes may be a source of at least a subpopulation of LP, but not spleen DCs, under steady-state conditions. In contrast, spleen and possibly LN conventional DCs develop from a separate BM-derived precursor population.100,102 One obvious question remaining is whether monocytes can only give rise to LP DCs under conditions of depletion, as occurs for Langerhans cells in the skin following ultraviolet irradiation, as no LP DCs were detectable without depletion.101 The fact the surface phenotype of the generated LP DCs was similar to what one finds in normal mice suggests that this may not be the case. While the progenitors for MLN DC populations have not been studied, these data suggest that conventional DCs in organized mucosal lymphoid tissues may be derived from different populations than LP DCs.55 Lastly, it was recently shown that some CD11chi as well as CD11cint cell populations from the small intestinal LP express F4/80.92 Whether these cell populations represent monocyte-derived cells is not at all clear, and raises caution in using the F4/80 as a marker of intestinal macrophages. Clearly more needs to be done to define the phenotype and ontological relationships of intestinal “DC” and “macrophage” populations.
Concluding Remarks
Recent studies point to a primary role for the local tissue environment in the conditioning of both DCs and macrophages in the steady-state to promote tissue specific immune responses that protect against pathology. In particular, factors produced by epithelial cells may be involved, as highlighted in several recent studies.103, 104, 105, 106 These may include TSLP (IL-50), TGFβ, and others produced under continuous signaling induced by intestinal flora. In addition, IL-10 from macrophages,87, 93 and DCs 69, 92 acting in an autocrine or paracrine manner, as well as prostaglandin E2 from stromal cells107 may significantly influence DC and macrophage function. The end result of such conditioning is to affect DCs to drive less pathological Th2 and Treg responses, and to positively affect IgA production against commensal organisms, as well as for macrophages to act as innate cells by phagocytosing and killing bacteria.
How microenvironental conditioning of DCs and TLR signaling can be overcome to initiate positive immune responses to pathogens is not yet clear. However, the use of virulence factors by pathogenic bacteria108 may induce the expression of chemokines, and inflammatory cytokines from epithelial109 or other cells, likely resulting in the recruitment of innate immune cells including neutrophils and macrophages, as well new DC precursors.110 Under these conditions, the change in the local milieu, including the production of IL-6, TNFα and IL1β would overcome regulatory effects of locally suppressive factors (TSLP, PGE2, and TGFβ) to drive monocyte differentiation into inflammatory macrophages and to activate new DC populations, which, following their migration to MLNs or to T-cell zones in mucosal follicles will drive effector rather than regulatory T-cell differentiation. In addition, the local production of proinflammatory factors (including IL-6) may subvert effector T-cell suppression by Tregs.111
Clearly a better understanding of how to define DC and macrophage populations in the intestine, and how they function together within local inductive and effector tissues has the potential to contribute greatly to the development of new vaccines and treatments for intestinal inflammation.
Disclosure
The authors declared no conflict of interest.
References
Kraehenbuhl, J.P. & Neutra, M.R. Epithelial M cells: differentiation and function. Annu. Rev. Cell Dev. Biol. 16, 301–332 (2000).
Owen, R.L. Sequential uptake of horseradish peroxidase by lymphoid follicle epithelium of Peyer's patches in the normal unobstructed mouse intestine: an ultrastructural study. Gastroenterology 72, 440–451 (1977).
Mantis, N.J. et al. Selective adherence of IgA to murine Peyer's patch M cells: evidence for a novel IgA receptor. J. Immunol. 169, 1844–1851 (2002).
Rey, J., Garin, N., Spertini, F. & Corthesy, B. Targeting of secretory IgA to Peyer's patch dendritic and T cells after transport by intestinal M cells. J. Immunol. 172, 3026–3033 (2004).
Corthesy, B. Roundtrip ticket for secretory IgA: role in mucosal homeostasis? J. Immunol. 178, 27–32 (2007).
Nochi, T. et al. A novel M cell-specific carbohydrate-targeted mucosal vaccine effectively induces antigen-specific immune responses. J. Exp. Med. 204, 2789–2796 (2007).
Chabot, S., Wagner, J.S., Farrant, S. & Neutra, M.R. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J. Immunol. 176, 4275–4283 (2006).
Chabot, S.M. et al. TLR2 activation by proteosomes promotes uptake of particulate vaccines at mucosal surfaces. Vaccine 25, 5348–5358 (2007).
Kelsall, B.L. & Strober, W. Distinct populations of dendritic cells are present in the subepithelial dome and T-cell regions of the murine Peyer's patch. J. Exp. Med. 183, 237–247 (1996).
Anosova, N.G. et al. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer's patches. Mucosal. Immunol. 1, 59–67 (2008).
Salazar-Gonzalez, R.M. et al. CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer's patches. Immunity 24, 623–632 (2006).
Iwasaki, A. & Kelsall, B.L. Localization of distinct Peyer's patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3alpha, MIP-3beta, and secondary lymphoid organ chemokine. J. Exp. Med. 191, 1381–1394 (2000).
Kunkel, D., Kirchhoff, D., Nishikawa, S., Radbruch, A. & Scheffold, A. Visualization of peptide presentation following oral application of antigen in normal and Peyer's patches-deficient mice. Eur. J. Immunol. 33, 1292–1301 (2003).
Hopkins, S.A. & Kraehenbuhl, J.P. Dendritic cells of the murine Peyer's patches colocalize with Salmonella typhimurium avirulent mutants in the subepithelial dome. Adv. Exp. Med. Biol. 417, 105–109 (1997).
Hopkins, S.A., Niedergang, F., Corthesy-Theulaz, I.E. & Kraehenbuhl, J.P. A recombinant Salmonella typhimurium vaccine strain is taken up and survives within murine Peyer's patch dendritic cells. Cell Microbiol. 2, 59–68 (2000).
Pron, B. et al. Dendritic cells are early cellular targets of Listeria monocytogenes after intestinal delivery and are involved in bacterial spread in the host. Cell Microbiol. 3, 331–340 (2001).
Salcedo, S.P. et al. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 4, e21 (2008).
Nagai, S. et al. Role of Peyer's patches in the induction of Helicobacter pylori-induced gastritis. Proc. Natl. Acad. Sci. USA 104, 8971–8976 (2007).
Fleeton, M.N. et al. Peyer's patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J. Exp. Med. 200, 235–245 (2004).
Macpherson, A.J. & Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665 (2004).
Gullberg, E. & Soderholm, J.D. Peyer's patches and M cells as potential sites of the inflammatory onset in Crohn's disease. Ann. NY Acad. Sci. 1072, 218–232 (2006).
Keita, A.V. et al. Characterization of antigen and bacterial transport in the follicle-associated epithelium of human ileum. Lab. Invest. 86, 504–516 (2006).
Turnbull, E.L., Yrlid, U., Jenkins, C.D. & Macpherson, G.G. Intestinal dendritic cell subsets: differential effects of systemic TLR4 stimulation on migratory fate and activation in vivo. J. Immunol. 174, 1374–1384 (2005).
Yoshida, M. et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Invest. 116, 2142–2151 (2006).
Rescigno, M. et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2, 361–367 (2001).
Niess, J.H. et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307, 254–258 (2005).
Chieppa, M., Rescigno, M., Huang, A.Y. & Germain, R.N. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 203, 2841–2852 (2006).
Brenchley, J.M. & Douek, D.C. HIV infection and the gastrointestinal immune system. Mucosal. Immunol. 1, 23–30 (2008).
Sansonetti, P.J. Rupture, invasion and inflammatory destruction of the intestinal barrier by Shigella: the yin and yang of innate immunity. Can. J. Infect. Dis. Med. Microbiol. 17, 117–119 (2006).
Karlsson, M. et al. “Tolerosomes” are produced by intestinal epithelial cells. Eur. J. Immunol. 31, 2892–2900 (2001).
Pugh, C.W., MacPherson, G.G. & Steer, H.W. Characterization of nonlymphoid cells derived from rat peripheral lymph. J. Exp. Med. 157, 1758–1779 (1983).
Huang, F.P. et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T-cell areas of mesenteric lymph nodes. J. Exp. Med. 191, 435–444 (2000).
Liu, L.M. & MacPherson, G.G. Lymph-borne (veiled) dendritic cells can acquire and present intestinally administered antigens. Immunology 73, 281–286 (1991).
Liu, L.M. & MacPherson, G.G. Antigen acquisition by dendritic cells: intestinal dendritic cells acquire antigen administered orally and can prime naive T cells in vivo. J. Exp. Med. 177, 1299–1307 (1993).
Worbs, T. et al. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J. Exp. Med. 203, 519–527 (2006).
Martinoli, C., Chiavelli, A. & Rescigno, M. Entry route of Salmonella typhimurium directs the type of induced immune response. Immunity 27, 975–984 (2007).
MacPherson, G.G., Jenkins, C.D., Stein, M.J. & Edwards, C. Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J. Immunol. 154, 1317–1322 (1995).
Yrlid, U. et al. Regulation of intestinal dendritic cell migration and activation by plasmacytoid dendritic cells, TNF-α and type 1 IFNs after feeding a TLR7/8 ligand. J. Immunol. 176, 5205–5212 (2006).
Uematsu, S. et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat. Immunol. 7, 868–874 (2006).
Hayashi, F. et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103 (2001).
Kelsall, B.L. & Leon, F. Involvement of intestinal dendritic cells in oral tolerance, immunity to pathogens, and inflammatory bowel disease. Immunol. Rev. 206, 132–148 (2005).
Johansson, C. & Kelsall, B.L. Phenotype and function of intestinal dendritic cells. Semin. Immunol. 17, 284–294 (2005).
Coombes, J.L. & Powrie, F. Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 8, 435–446 (2008).
Iwasaki, A. Mucosal dendritic cells. Annu. Rev. Immunol. 25, 381–418 (2007).
Campbell, D.J. & Butcher, E.C. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J. Exp. Med. 195, 135–141 (2002).
Johansson-Lindbom, B. et al. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J. Exp. Med. 198, 963–969 (2003).
Svensson, M. et al. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J. Clin. Invest. 110, 1113–1121 (2002).
Mora, J.R. et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature 424, 88–93 (2003).
Stagg, A.J., Kamm, M.A. & Knight, S.C. Intestinal dendritic cells increase T-cell expression of alpha4beta7 integrin. Eur. J. Immunol. 32, 1445–1454 (2002).
Mora, J.R. et al. Reciprocal and dynamic control of CD8 T cell homing by dendritic cells from skin- and gut-associated lymphoid tissues. J. Exp. Med. 201, 303–316 (2005).
Iwata, M. et al. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538 (2004).
Saurer, L., McCullough, K.C. & Summerfield, A. In vitro induction of mucosa-type dendritic cells by all-trans retinoic acid. J. Immunol. 179, 3504–3514 (2007).
Annacker, O. et al. Essential role for CD103 in the T-cell-mediated regulation of experimental colitis. J. Exp. Med. 202, 1051–1061(2005).
Johansson-Lindbom, B. et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T-cell homing. J. Exp. Med. 202, 1063–1073 (2005).
Svensson, M. et al. Retinoic acid receptor signaling levels and antigen dose regulate gut homing receptor expression on CD8+ T cells. Mucosal. Immunol. 1, 38–48 (2008).
Mora, J.R. et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006).
Suzuki, K., Ha, S.A., Tsuji, M. & Fagarasan, S. Intestinal IgA synthesis: a primitive form of adaptive immunity that regulates microbial communities in the gut. Semin. Immunol. 19, 127–135 (2007).
Macpherson, A.J. IgA adaptation to the presence of commensal bacteria in the intestine. Curr. Top. Microbiol. Immunol. 308, 117–136 (2006).
Macpherson, A.J. et al. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288, 2222–2226 (2000).
Becker, C. et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J. Clin. Invest. 112, 693–706 (2003).
Sato, A. et al. CD11b+ Peyer's patch dendritic cells secrete IL-6 and induce IgA secretion from naive B cells. J. Immunol. 171, 3684–3690 (2003).
Faria, A.M. & Weiner, H.L. Oral tolerance. Immunol. Rev. 206, 232–259 (2005).
Dubois, B., Goubier, A., Joubert, G. & Kaiserlian, D. Oral tolerance and regulation of mucosal immunity. Cell Mol. Life Sci. 62, 1322–1332 (2005).
Faria, A.M. & Weiner, H.L. Oral tolerance: therapeutic implications for autoimmune diseases. Clin. Dev. Immunol. 13, 143–157 (2006).
Zhang, X., Izikson, L., Liu, L. & Weiner, H.L. Activation of CD25+CD4+ regulatory T cells by oral antigen administration. J. Immunol. 167, 4245–4253 (2001).
Thorstenson, K.M. & Khoruts, A. Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J. Immunol. 167, 188–195 (2001).
Dubois, B. et al. Innate CD4+CD25+ regulatory T cells are required for oral tolerance and inhibition of CD8+ T cells mediating skin inflammation. Blood 102, 3295–3301 (2003).
Viney, J.L., Mowat, A.M., O'Malley, J.M., Williamson, E. & Fanger, N.A. Expanding dendritic cells in vivo enhances the induction of oral tolerance. J. Immunol. 160, 5815–5825 (1998).
Iwasaki, A. & Kelsall, B.L. Freshly isolated Peyer's patch, but not spleen, dendritic cells produce interleukin 10 and induce the differentiation of T helper type 2 cells. J. Exp. Med. 190, 229–239 (1999).
Alpan, O., Rudomen, G. & Matzinger, P. The role of dendritic cells, B cells, and M cells in gut-oriented immune responses. J. Immunol. 166, 4843–4852 (2001).
Iwasaki, A. & Kelsall, B.L. Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer's patch dendritic cells. J. Immunol. 166, 4884–4890 (2001).
Chirdo, F.G., Millington, O.R., Beacock-Sharp, H. & Mowat, A.M. Immunomodulatory dendritic cells in intestinal lamina propria. Eur. J. Immunol. 35, 1831–1840 (2005).
Coombes, J.L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-β and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Mucida, D. et al. Reciprocal TH17 and regulatory T-cell differentiation mediated by retinoic acid. Science 317, 256–260 (2007).
Sun, J.C., Williams, M.A. & Bevan, M.J. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 5, 927–933 (2004).
Sakaguchi, S. et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol. Rev. 212, 8–27 (2006).
Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006).
Chen, W. et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 (2003).
Fantini, M.C. et al. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 172, 5149–5153 (2004).
Fantini, M.C. et al. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 55, 671–680 (2006).
Sun, C.M. et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785 (2007).
Yamazaki, S. et al. Dendritic cells are specialized accessory cells along with TGF-{beta} for the differentiation of Foxp3+ CD4+ regulatory T cells from peripheral Foxp3− precursors. Blood 110, 4293–4302 (2007).
Schambach, F., Schupp, M., Lazar, M.A. & Reiner, S.L. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur. J. Immunol. 37, 2396–2399 (2007).
Benson, M.J., Pino-Lagos, K., Rosemblatt, M. & Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 204, 1765–1774 (2007).
Lee, S.H., Starkey, P.M. & Gordon, S. Quantitative analysis of total macrophage content in adult mouse tissues. Immunochemical studies with monoclonal antibody F4/80. J. Exp. Med. 161, 475–489 (1985).
Hume, D.A., Loutit, J.F. & Gordon, S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80: macrophages of bone and associated connective tissue. J. Cell Sci. 66, 189–194 (1984).
Hirotani, T. et al. The nuclear IkappaB protein IkappaBNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. J. Immunol. 174, 3650–3657 (2005).
Rogler, G. et al. Isolation and phenotypic characterization of colonic macrophages. Clin. Exp. Immunol. 112, 205–215 (1998).
Smythies, L.E. et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J. Clin. Invest. 115, 66–75 (2005).
Leon, F., Smythies, L.E., Smith, P.D. & Kelsall, B.L. Involvement of dendritic cells in the pathogenesis of inflammatory bowel disease. Adv. Exp. Med. Biol. 579, 117–132 (2006).
Platt, A.M. & Mowat, A.M. Mucosal macrophages and the regulation of immun responses in the intestine. Immunol. Lett. 119, 22–31 (2008).
Monteleone, I., Platt, A.M., Jaensson, E., Agace, W.W. & Mowat, A.M. IL-10-dependent partial refractoriness to Toll-like receptor stimulation modulates gut mucosal dendritic cell function. Eur. J. Immunol. 38, 1533–1547 (2008).
Denning, T.L., Wang, Y.C., Patel, S.R., Williams, I.R. & Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 8, 1086–1094 (2007).
Kamada, N. et al. Abnormally differentiated subsets of intestinal macrophage play a key role in Th1-dominant chronic colitis through excess production of IL-12 and IL-23 in response to bacteria. J. Immunol. 175, 6900–6908 (2005).
Wick, M.J. Monocyte and dendritic cell recruitment and activation during oral Salmonella infection. Immunol. Lett. 112, 68–74 (2007).
Serbina, N.V., Salazar-Mather, T.P., Biron, C.A., Kuziel, W.A. & Pamer, E.G. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 19, 59–70 (2003).
Tezuka, H. et al. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 448, 929–933 (2007).
Strauss-Ayali, D., Conrad, S.M. & Mosser, D.M. Monocyte subpopulations and their differentiation patterns during infection. J. Leukoc. Biol. 82, 244–252 (2007).
Geissmann, F., Jung, S. & Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82 (2003).
Fogg, D.K. et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 311, 83–87 (2006).
Varol, C. et al. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 204, 171–180 (2007).
Naik, S.H. et al. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat. Immunol. 7, 663–671 (2006).
Rescigno, M. & Chieppa, M. Gut-level decisions in peace and war. Nat. Med. 11, 254–255 (2005).
Rimoldi, M. et al. Intestinal immune homeostasis is regulated by the crosstalk between epithelial cells and dendritic cells. Nat. Immunol. 6, 507–514 (2005).
Zaph, C. et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature 446, 552–556 (2007).
Nenci, A. et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561 (2007).
Newberry, R.D., McDonough, J.S., Stenson, W.F. & Lorenz, R.G. Spontaneous and continuous cyclooxygenase-2-dependent prostaglandin E2 production by stromal cells in the murine small intestine lamina propria: directing the tone of the intestinal immune response. J. Immunol. 166, 4465–4472 (2001).
Hacker, J. & Kaper, J.B. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679 (2000).
Kim, J.M. et al. Apoptosis of human intestinal epithelial cells after bacterial invasion. J. Clin. Invest. 102, 1815–1823 (1998).
Iliev, I.D., Matteoli, G. & Rescigno, M. The yin and yang of intestinal epithelial cells in controlling dendritic cell function. J. Exp. Med. 204, 2253–2257 (2007).
Pasare, C. & Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036 (2003).
Mowat, A.M. et al. The role of dendritic cells in regulating mucosal immunity and tolerance. Novartis. Found. Symp. 252, 291–302 (2003).
Vremec, D. & Shortman, K. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159, 565–573 (1997).
Yrlid, U. & Wick, M.J. Antigen presentation capacity and cytokine production by murine splenic dendritic cell subsets upon Salmonella encounter. J. Immunol. 169, 108–116 (2002).
Sundquist, M. & Wick, M.J. TNF-alpha-dependent and -independent maturation of dendritic cells and recruited CD11c(int)CD11b+ Cells during oral Salmonella infection. J. Immunol. 175, 3287–3298 (2005).
Karlis, J. et al. Characterization of colonic and mesenteric lymph node dendritic cell subpopulations in a murine adoptive transfer model of inflammatory bowel disease. Inflamm. Bowel. Dis. 10, 834–847 (2004).
Mottet, C., Uhlig, H.H. & Powrie, F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170, 3939–3943 (2003).
Uematsu, S. et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 9, 769–776 (2008).
Becker, C. et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J. Clin. Invest. 112, 693–706 (2003).
Vallon-Eberhard, A., Landsman, L., Yogev, N., Verrier, B. & Jung, S. Transepithelial pathogen uptake into the small intestinal lamina propria. J. Immunol. 176, 2465–2469 (2006).
Asselin-Paturel, C., Brizard, G., Pin, J.-J., Briere, F. & Trinchieri, G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J. Immunol. 171, 6466–6477 (2003).
Bilsborough, J., George, T.C., Norment, A. & Viney, J.L. Mucosal CD8alpha+ DC, with a plasmacytoid phenotype, induce differentiation and support function of T cells with regulatory properties. Immunology 108, 481–492 (2003).
Castellaneta, A., Abe, M., Morelli, A.E. & Thomson, A.W. Identification and characterization of intestinal Peyer's patch interferon-alpha producing (plasmacytoid) dendritic cells. Hum. Immunol. 65, 104–113 (2004).
Contractor, N., Louten, J., Kim, L., Biron, C. & Kelsall, B.L. Peyer's patch plasmacytoid dendritic cells (pDC) produce low levels of type I interferon; possible role for IL-10, TGFβ and PGE2 in conditioning a unique mucosal pDC phenotype. J. Immunol. 179, 2690–2694 (2007).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Kelsall, B. Recent progress in understanding the phenotype and function of intestinal dendritic cells and macrophages. Mucosal Immunol 1, 460–469 (2008). https://doi.org/10.1038/mi.2008.61
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2008.61
This article is cited by
-
Host–microbiota interactions in inflammatory bowel disease
Nature Reviews Immunology (2020)
-
CCR2+CD103− intestinal dendritic cells develop from DC-committed precursors and induce interleukin-17 production by T cells
Mucosal Immunology (2015)
-
Dendritic cells of the oral mucosa
Mucosal Immunology (2014)
-
Contributions of dendritic cells and macrophages to intestinal homeostasis and immune defense
Immunology & Cell Biology (2013)
-
Cross-presentation of IgG-containing immune complexes
Cellular and Molecular Life Sciences (2013)
