
Abstract
The classical transmission disequilibrium test (TDT) based on a trio design uses information only on the allele transmitted by a heterozygous parent at a marker locus as homozygous parents are non-informative about linkage. However, the phenotype of an offspring depends on the alleles transmitted by both parents, irrespective of whether the parents are homozygous or heterozygous, and hence carry useful information on association. In this article, we propose modifications to the TDT procedures by incorporating transmission data on both parents in an informative trio to explore possible gain in power in detecting association. For a binary trait, we use a goodness-of-fit χ2 test, whereas for a quantitative trait, we devise two tests: one based on a bivariate response logistic model and the other using a quasi-likelihood approach. We evaluate the type 1 errors and the powers of the proposed tests with those of the classical TDT procedures for both binary and quantitative traits based on extensive simulations. We find that the inclusion of transmission data on non-informative parents yields marginally higher power in the logistic regression approach but results in substantial gain in power in the quasi-likelihood approach. We apply our proposed methods to analyze a count phenotype related to alcoholism.
Similar content being viewed by others


Introduction
The classical transmission disequilibrium test (TDT) for binary traits1 based on a trio design is a popular family-based alternative to population-based case–control studies as it tests for association in the presence of linkage, and hence is protected against population stratification. As many binary clinical end-point traits are governed by one or more quantitative precursors (e.g., homocysteine levels and vitamin B12 levels are precursors of coronary artery disease), association analyses of these quantitative traits have also been an active area of research. Although several model-based tests for transmission disequilibrium have been developed for quantitative traits,2, 3, 4 the test proposed by Waldman et al.5 and modified by Haldar and Ghosh6 are based on the logistic regression framework, which uses similar approach as the classical TDT and do not require any distributional assumption on quantitative trait values. However, most existing test procedures to detect transmission disequilibrium for both binary and quantitative traits use data only on those parents who are heterozygous at the marker locus of interest, as informations on linkage are not contained in allelic transmissions by homozygous parents. On the other hand, the phenotype of an individual is a function of the genotype of the individual at a trait locus, implying that useful information on genetic association are contained in the alleles transmitted by both parents of the individual, irrespective of whether the other parent is homozygous or heterozygous. Ghosh and Reich7 had explored the effect of including homozygous parents in the classical TDT framework in which they had considered the frequencies of the different combinations of transmission and non-transmission of the two alleles at the marker locus and constructed a χ2 test statistic based on the deviation of these observed frequencies from those expected under the null hypothesis of no association. The power of the test was lower than the classical TDT as the deviations corresponding to homozygous parents are smaller compared with heterozygous parents. However, Terwillger and Ott8 derived a haplotype-based relative risk statistic haplotype-based haplotype relative risk (HHRR) where the variance of the difference in transmission frequencies is estimated using data from all parents and showed that it provides higher power than the classical TDT under certain disease models.
Hence, it is of interest to develop statistical tests that incorporate data on allelic transmissions from both parents (subject to at least one of them being heterozygous) to an offspring and explore whether the inclusion of transmission information from homozygous parents in such analyses results in increased powers of the tests.
In this article, we explore some novel statistical methods that use information on the allelic transmissions from both parents to an offspring in a nuclear family for detecting genetic association with respect to binary as well as quantitative traits. For binary traits, we consider a goodness-of-fit χ2 test for the joint distribution of the allelic transmissions from the two parents to an affected offspring. For quantitative traits, we explore a bivariate response logistic model of the parental allelic transmissions conditioned on the quantitative trait value of the offspring. We also consider a competing approach based on a quasi-likelihood framework that models the differences between the allelic transmissions of the two parents and their expected values conditioned on the quantitative trait value of the offspring. We carry out extensive simulations under a wide spectrum of genetic models and different probability distributions of quantitative trait values to assess the type 1 errors and compare the powers of the proposed tests. We also illustrate an application of the methods using data on externalizing symptoms, an alcohol-related endophenotype from the Collaborative Study on the Genetics of Alcoholism.
Materials and methods
We assume that a binary disease trait (1, if affected and 0, otherwise) and a quantitative trait Y are both controlled by an autosomal biallelic locus with alleles A and a with minor allele frequency P(A)=q. We consider a biallelic marker locus with allele M1 and M2 with minor allele frequency P(M1)=m. The coefficient of linkage disequilibrium between the minor alleles at the two loci is denoted by δ. Suppose the penetrance of the disease corresponding to the genotypes at the trait locus AA, Aa and aa are f2, f1 and f0, respectively, while for quantitative traits, f2, f1 and f0 denote the probability density functions of Y conditioned on the three genotypes. We assume that the allele A increases the risk of the disease or pertains to higher values of the quantitative trait Y. For the binary trait, we assume that marker genotype data are available on trios ascertained through an affected offspring and satisfying the condition that at least one parent is heterozygous at the marker locus. For the quantitative trait, we consider marker genotype data on trios (two parents and an offspring) such that at least one parent is heterozygous at the marker locus along with quantitative trait data on the offspring.
We define Z1 and Z2 to be indicator variables corresponding to the transmission of the allele M1 by the heterozygous parent and the other parent, respectively. If both the parents are heterozygous at the marker locus, then we assign one transmission as Z1 and other as Z2 randomly. We note that the identification of Z1 and Z2 are not affected if the offspring is homozygous. On the other hand, while Z1 and Z2 cannot be identified for a heterozygous offspring, P(Z1=1, Z2=0) and P(Z1=0, Z2=1) are equally likely in the absence of association. Haldar and Ghosh6 showed that if only one transmission from such families are considered, the test for association maintains the correct size. Suppose denotes the conditional probability of Z1=i and Z2=j given the phenotype value (affected in case of binary trait) i, j=0,1 and l=0,1,2 denote the number of minor allele of the genotype at the trait locus, then under the complex disease model, the joint distribution of Z1 and Z2 is given as

where Ψlij=P(Z1=i, Z2=j|l) denotes the conditional probabilities of Z1 and Z2 given that l is the number of minor allele of the genotype at trait loci (the expressions are provided in Supplementary Equation S1), pl, l=0,1,2 denotes the genotype frequencies at the quantitative trait loci (QTL) and

is the prevalence of the disease (unconditional probability density function of trait values for quantitative traits) in the population. Under the null hypothesis of no association in the presence of linkage, the null distribution of Z1 and Z2 is given by

Thus, testing the null hypothesis of no association is equivalent to testing π00=π10 and π01=π11. The proof of the above equivalence is provided in Supplementary Equation S1. It can be easily deduced from Supplementary Equations S1.1 and S1.2 that π10–π00>0 and π11–π01>0 if δ>0, whereas both the quantities are negative if δ<0.
Binary traits
The classical TDT statistic1 based on the trio framework as described above is given by

where  and
and  are the total number of heterozygous parents transmitting the alleles M1 and M2, respectively, to their offspring. Here we extend the above test procedure by including the transmission of the other parents. Let nij be the number of observation for which Z1=i and Z2=j and i,j=0,1, then the hypothesis given in (Supplementary Equation S1) can tested using the goodness-of-fit test statistic as
are the total number of heterozygous parents transmitting the alleles M1 and M2, respectively, to their offspring. Here we extend the above test procedure by including the transmission of the other parents. Let nij be the number of observation for which Z1=i and Z2=j and i,j=0,1, then the hypothesis given in (Supplementary Equation S1) can tested using the goodness-of-fit test statistic as

where eij is the expected value of nij under the null hypothesis. On simplification, the above test statistic can be expressed as

and is asymptotically distributed as χ2 with 2 degrees of freedom.
Quantitative traits
Waldman et al.5 proposed a test based on a logistic regression framework that models the probability of transmission of the allele M1 from a heterozygous parent to a offspring conditioned on the offspring’s quantitative trait value. The model is given by

where z denotes the indicator for the transmission of allele M1 by a heterozygous parent. Haldar and Ghosh6 proposed a modified logistic regression model that does not contain any intercept parameter and is conditioned on the mean-adjusted quantitative trait values of the offspring as follows:

The test for association in the presence of linkage transmission-based association test (TBAT)6 is based on the parameter β and yields comparable power as the popular approach family-based association test (FBAT),9 but is computationally much less expensive.
We model the joint distribution of the alleles transmitted by the two parents using a logistic regression with two responses:10

The null hypothesis of no linkage or no association is equivalent to β1=β2=β3=0. Thus, a TDT can be constructed using likelihood ratio principles and is given by

where l denotes the log-likelihood function, α0 and α1 are the maximum-likelihood estimates of α under the null and the alternative hypothesis, respectively, whereas β1, β2 and β3 are the maximum-likelihood estimates of β1, β2 and β3, respectively. The test statistic is asymptotically distributed as  under the null hypothesis.
under the null hypothesis.
An alternative test statistic can be defined as

The false discovery rates corresponding to the above test statistic is obtained using the Benjamini–Hochberg multiple testing correction.11
We also adopt the quasi-likelihood approach proposed by Wedderburn12 based on generalized linear models given by McCullagh and Nelder13 to develop a competing test for transmission disequilibrium. In the trio framework, the quasi-likelihood equation is given by

where Zi=(z1i, z2i) is the transmission vector for the ith family, z1i is the indicator for the transmission of the allele M1 by the heterozygous parent and z2i is the indicator for the transmission of the allele M1 by the other parent. If both parents are heterozygous, then we assign the transmission of one parent as z1i and other as z2i randomly, so that every family has single observation. Let μi=(μ1i, μ2i)=(E(z1i|yi), E(z2i|yi)) and Vi=V(Zi|y) be the conditional variance–covariance matrix of Zi. We use a logistic link function corresponding to the mean as follows:

For simplicity, we assume that Vi is the same for all i and equal to V.
We note that for large values of yi (i.e.,  ) β>0 implies that E(z1i|yi)>0.5, whereas β<0 implies that E(z1i|yi)<0.5. The implications are in the reverse directions for small values of yi (i.e.,
) β>0 implies that E(z1i|yi)>0.5, whereas β<0 implies that E(z1i|yi)<0.5. The implications are in the reverse directions for small values of yi (i.e.,  ).
).
The parameter γ=(α, β) can be estimated using the generalized estimating equations (GEE)14, 15

The GEE procedure is known to be highly influenced by the presence of outliers. As extreme phenotype values are common in association studies, the above GEE approach may not be an efficient strategy. Thus, we assume a modified resistance GEE16, 17 model given as follows:

where Wi is a function that downweights the extreme values. We use the weight function  , where
, where  ,
,  and
and  . Using the Taylor series expansion on Equation (1) around the true parameter γ, we obtain
. Using the Taylor series expansion on Equation (1) around the true parameter γ, we obtain

where  . The variance of γ can be estimated using the robust sandwich estimator as follows:
. The variance of γ can be estimated using the robust sandwich estimator as follows:

where  is the estimated residual vector and the variance–covariance matrix of the transmission vector is estimated using the plug in estimator given by
is the estimated residual vector and the variance–covariance matrix of the transmission vector is estimated using the plug in estimator given by

We note that in Equation (2)

and V(β) is the (2, 2)th element of V(γ).
The null hypothesis of no association or no linkage is equivalent to β=0 and the test statistic is given by

and is asymptotically distributed as  under the null.
under the null.
Results
Simulation study
To compare TDTB with the classical TDT1 as well as TBATBL,TBATMax and TBATQL with TBAT with respect to power of detecting association in the presence of linkage, we carried out extensive simulations under a wide spectrum of genetic models and probability distributions of the underlying trait. We generate genotype data for three choices of minor allele frequencies: 0.1, 0.2 and 0.3 for both the trait and the marker loci. We consider different models of disease inheritance: recessive, dominant (both with incomplete penetrances) and complex such that the prevalence of the underlying disease is ∼10% under each of the models. We fix the value of the recombination fraction θ between the two loci as 0.01 and consider four different levels for linkage disequilibrium δ=0 (no association), 0.33, 0.67 and 1 (complete association). (The details on the simulation parameters considered under the different models are provided in Supplementary Equation S2).
For quantitative traits, we use the different combination of the values 0.1, 0.3 and 0.5 as minor allele at the marker and QTL. Assuming that A and a are the two alleles at the QTL, the conditional mean and variance of the trait values given the genotypes AA, Aa and aa are μ2, μ1, μ0 and σ2, σ1, σ0, respectively. The trait values are generated using different probability models: normal (reflecting symmetric QT distributions), location- and scale-shifted χ2 (reflecting skewed QT distributions) and location-shifted Poisson (reflecting count phenotypes) by varying the means and variances of the quantitative traits conditioned on QTL genotypes. (The details on the simulation parameters considered under the different models are provided in Supplementary Equation S3).
In the case of binary traits (results are provided in Table 1), our simulations reveal that the power of the proposed test procedure TDTB is an increasing function of the heterozygosity at the disease locus for recessive models of inheritance and is a decreasing function of heterozygosity at the disease locus for dominant and complex models. However, the classical TDT statistic yields more power compared with TDTB. On the other hand, the rate of increase in power with increase in the coefficient of linkage disequilibrium is comparatively more for the TDTB procedure.
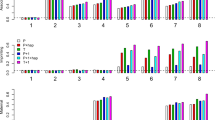
For quantitative traits, we observe that under certain scenarios, the test statistics based on the bivariate logistic model TBATBL and TBATMax yield marginally higher rates of false positives than the desired level of 0.05, whereas TBATQL maintains the desired level for all parameters considered (Figure 1). We also observe that the powers corresponding to TBATBL and TBATMax are marginally higher than TBAT (that considers transmission data only from heterozygous parents) if the minor allele frequency at the marker locus is high but yields comparable power as TBAT for smaller values of minor allele frequency. On the other hand, the quasi-likelihood approach TBATQL produces consistently higher powers compared with TBAT, TBATBL and TBATMax. We note that TBATBL, is a χ2 test with 3 degrees of freedom, whereas TBATQL is a χ2 test with 1 degree of freedom. It is likely that the additional degrees of freedom corresponding to TBATBL results in a reduction in power compared with TBATQL. The simulations also reveal that TBATMax yields more power compared with TBATBL (as evident from Figure 2). All the test procedures give similar powers across different probability distributions of quantitative trait values (comparing Figures 2,3,4), and hence is robust to possible violations in underlying distributional assumptions. We also observe that the powers of the all test procedures increases with the extent of dominance (Figure 5) and decreases with the extent of heteroskedasticity across the genotypes at the QTL (Figure 6). We also found that under population stratification, the modified approach that includes information on transmission of both parents is susceptible to inflated rates of type I errors (results not shown for brevity).

Empirical type 1 error rates corresponding to the different test statistics when the quantitative trait values are distributed as normal.

Empirical powers corresponding to the different test statistics when the quantitative trait values are distributed as normal under an additive model.

Empirical powers corresponding to the different test statistics when the quantitative trait values are distributed as location- and scale-shifted χ2 under an additive model.

Empirical powers corresponding to the different test statistics when the quantitative trait values are distributed as location-shifted Poisson under an additive model.

Empirical powers corresponding to the different test statistics when the quantitative trait values are distributed as normal under a dominance model.

Empirical powers corresponding to the different test statistics when the quantitative trait values are distributed as normal under a heteroskedasticity model.
An application to Collaborative Study on the Genetics of Alcoholism phenotype
The Collaborative Study on the Genetics of Alcoholism is a multicenter research initiative with the objective of identifying susceptibility genes for alcohol dependence and related phenotypes. There is increasing evidence that externalizing behavior disorders such as conduct disorder are strongly correlated with alcohol use disorders. Ghosh et al.18 performed a genome-wide linkage scan on a quantitative endophenotype defined as the number of externalizing symptoms related to antisocial behavioral traits. The phenotype is the count of 24 symptoms endorsed by an individual and hence ranges between 0 and 24. Multipoint nonparametric regression based on 171 independent sib-pairs revealed significant evidence of linkage in the 4q22.3 region on chromosome 4 harboring the alcohol dehydorogenase gene cluster. One of the linkage markers genotyped in this region was a biallelic marker ADH1C. We applied the proposed bivariate logistic regression (TBATBL) and the quasi-likelihood approach (TBATQL) on 138 independent trios with at least one parent heterozygous at the locus. Although both the procedures provided significant evidence of over transmission of one of the alleles compared with the other allele for high values of the quantitative endophenotype, indicating that the marker locus may be in strong linkage disequilibrium with a QTL modulating the number of externalizing symptoms, the significance provided by TBATQL (P-value <0.00001) was much more pronounced compared with TBATBL (P-value <0.0001).
Discussion
The classical TDT1 for association in the presence of linkage is based on a trio design and uses information on allelic transmission only from heterozygous parents. As the phenotype of an offspring depends on his/her genotype at the trait locus, and hence on the combination of alleles inherited from the two parents, information on genetic association is provided not only by the allele transmitted by a heterozygous parent in a trio but also by the allele transmitted by the other parent, irrespective of whether that parent is homozygous or heterozygous. Terwillger and Ott8 and Ghosh and Reich7 had studied the effect of including transmission information from homozygous parents in the classical TDT framework in which they had considered transmission data from the two parents within a trio to be independent of each other and had obtained contrasting results with respect to powers of their proposed tests vis-a-vis the classical TDT.
The purpose of this article was to explore the effect of including data on allelic transmissions simultaneously from both parents to an offspring in the framework of the classical TDT design for binary as well as quantitative traits. Our proposed joint modeling of the transmitted alleles from the two parents in an informative trio to an affected offspring (TDTB) does not provide any advantage and yields lower power in detecting association compared with the classical TDT that is based on transmissions only from heterozygous parents. On the other hand, the bivariate logistic regression approach (TBATBL) provides marginal increase in power compared with FBAT9 and TBAT6 with respect to association tests for quantitative traits. However, the proposed quasi-likelihood approach (TBATQL) based on resistance GEE provides a significant gain in power over the other test procedures, while maintaining the model free property of TDT statistics. While the modified test procedure has higher power compared with TBAT,6 it is important to note that, unlike TBAT, the test is not protected against population stratification, and hence the power comparison needs to be restricted to genetically homogeneous populations.
References
Spielman, R. S., McGinnis, R. E. & Ewens, W. J. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Ann. J. Hum. Genet. 52, 508–516 (1993).
Allison, D. B. Transmission-disequilibrium tests for quantitative traits. Am. J. Hum. Genet. 60, 676–690 (1997).
George, V., Tiwari, H. K., Shu, Y., Zhu, X. & Elston, R. C. Linkage and association analysis of alcoholism using a regression-based transmission/disequilibrium test. Genet. Epidemiol. 17 (suppl), 157–161 (1999).
Abecasis, G. R., Cardon, L. R. & Cookson, W. O. A general test of association for quantitative traits in nuclear families. Am. J. Hum. Genet. 66, 279–292 (2000).
Waldman, I. D., Robinson, B. F. & Rowe, D. C. A logistic regression based extension of the TDT for continuous and categorical traits. Ann. Hum. Genet. 63, 329–340 (1999).
Haldar, T. & Ghosh, S. Statistical equivalent of the classical TDT for quantitative traits and multivariate phenotypes. J. Genet. 94, 619–628 (2015).
Ghosh, S. & Reich, T. How do homozygous parents affect TDT as a test for association. Hum. Hered. 53, 181–186 (2002).
Terwillger, J. D. & Ott, J. A haplotype-based “haplotype relative risk” approach to detecting allelic associations. Hum. Hered. 42, 337–346 (1992).
Lange, C. & Laird, N. M. On a general class of conditional tests for family-based association studies in genetics: the asymptotic distribution,the conditional power, and optimality considerations. Genet. Epidemiol. 23, 165–180 (2002).
Cox, D. R. & Snell, E. J. Analysis of Binary Data, 2nd edn (Chapman & Hall/CRC Press, London/New York, 1989).
Benjamini, Y. & Hocheberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B 57, 289–300 (1995).
Wedderburn, R. W. M. Quasi-likelihood functions, generalized linear models, and the Gauss–Newton method. Biometrika 61, 439–447 (1974).
McCullagh, P. & Nelder, J. A. Generalized Linear Models, (Chapman & Hall, London, 1989).
Gourieroux, C., Monfort, A. & Trognon, A. Pseudo maximum likelihood methods: applications to Poisson models. Econometrica 52, 701–720 (1984).
Liang, K. Y. & Zeger, S. L. Longitudinal data analysis using generalized linear models. Biometrika 73, 13–22 (1986).
Preisser, J. S. & Qaqish, B. F. Robust regression for clustered data with applications to binary regression. Biometrics 55, 574–579 (1999).
Eva, C. & Elvezio, R. Robust inference for generalized linear models. JASA Theory Method 96, 1022–1030 (2001).
Ghosh, S., Bierut, L. J., Porjesz, B., Edenberg, H. J., Dick, D., Goate, A. et al. A novel non-parametric regression reveals linkage on chromosome 4 for the number of externalizing symptoms in sib-pairs. Am. J. Med. Genet. B 147B, 1301–1305 (2008).
Acknowledgements
This work was partially supported by the Senior Research Fellowship of the Indian Statistical Institute to Hemant Kulkarni. We are grateful to Professor Laura J Bierut for providing access to the data on externalizing symptoms generated in the Collaborative Study on the Genetics of Alcoholism project. Both the authors have contributed to the development of statistical methodology and preparing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Kulkarni, H., Ghosh, S. Including non-informative parents in transmission-based association tests. J Hum Genet 62, 621–629 (2017). https://doi.org/10.1038/jhg.2017.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.9
