
Abstract
Mutations in the fukutin-related protein (FKRP) gene have been associated with dystroglycanopathies, which are common in Europe but rare in Asia. Our study aimed to retrospectively analyze and characterize the clinical, myopathological and genetic features of 12 Chinese patients with FKRP mutations. Three patients were diagnosed with congenital muscular dystrophy type 1C (MDC1C) and nine patients were diagnosed with limb girdle muscular dystrophy type 2I (LGMD2I). Three muscle biopsy specimens had dystrophic changes and reduced glycosylated α-dystroglycan staining, and two showed reduced expression of laminin α2. Two known and 13 novel mutations were identified in our single center cohort. Interestingly, the c.545A>G mutation was found in eight of the nine LGMD2I patients as a founder mutation and this founder mutation in Chinese patients differs from the one seen in European patients. Moreover, patients homozygous for the c.545A>G mutation were clinically asymptomatic, a less severe phenotype than in compound heterozygous patients with the c.545A>G mutation. The 13 novel mutations of FKRP significantly expanded the mutation spectrum of MDC1C and LGMD2I, and the different founder mutations indicate the ethnic difference in FKRP mutations.
Similar content being viewed by others

Introduction
Mutations in the fukutin-related protein (FKRP) gene are associated with the dystroglycanopathies, which are caused by glycosylation defects of α-dystroglycan (α-DG).1 Mutations in FKRP are responsible for several phenotypes of dystroglycanopathies, such as Walker–Warburg syndrome, muscle–eye–brain disease, congenital muscular dystrophy type 1C (MDC1C) and limb girdle muscular dystrophy type 2I (LGMD2I).2, 3 LGMD2I is one of the most common dystroglycanopathies in Europe but is rarely reported in Asia. To date, only 10 cases of LGMD2I have been reported in Asians, including 6 from Taiwan,4 2 from mainland China,5 1 from Japan4 and 1 Asian-American from North America.6 Here we report clinical, myopathological and genetic study findings from 12 Chinese patients carrying FKRP mutations, including 3 patients with MDC1C and 9 with LGMD2I.
Materials and methods
Patients
Twelve Chinese patients carrying FKRP mutations were recruited from Peking University First Hospital between 2007 and 2016. The patients (7 males, 5 females) from 12 non-consanguineous families were all Han Chinese, from 9 provinces of China. The research protocol was reviewed and approved by the Ethics Committee of the Peking University First Hospital (Beijing, China). Written informed consent was obtained to participate in the study, as well as publish medical data from controls, all patients and/or their parents. Clinical data were collected (Table 1), including sex, age, age of onset, initial symptoms, motor development, mental development, pattern of muscle involvement, joint contracture, creatine kinase (CK) level in serum, muscle pathology or muscle magnetic resonance imaging (MRI) and brain involvement.
Muscle biopsy
Open muscle biopsy of the quadriceps femoris muscle was carried out on three patients. Biopsied muscle specimens were frozen in isopentane cooled in liquid nitrogen. Frozen sections (8 μm) were processed. Serial frozen sections were stained with hematoxylin and eosin. Immunohistochemical and immunofluorescent staining was performed with α-DG mouse monoclonal antibody, clone IIH6C4 (1:20, 40 μl; no. 05-593; immunogen, rabbit skeletal muscle membrane preparation; Merck Millipore, Darmstadt, Germany), and laminin α2 mouse monoclonal antibody, clone 5H2 (1:20, 40 μl; no. MAB1922; immunogen, purified human merosin; Merck Millipore).
PCR and direct sequencing
Genomic DNA was extracted from anticoagulated whole blood using the Qiagen Blood DNA Extraction Kit (Qiagen, Hilden, Germany). PCR and direct sequencing was performed to find the mutation and to confirm mutations found by next-generation sequencing. The open reading frame of FKRP (NM_001039885) was PCR-amplified using oligonucleotide primers designed using Primer 5.0 (Premier Biosoft, Palo Alto, CA, USA). The PCR products were purified and directly sequenced using an automatic sequencer (ABI PRISM 3730 XL; Applied Biosystems, Foster City, CA, USA). Sequences were analyzed using the DNASTAR software (Madison, WI, USA).
Next-generation sequencing
Next-generation sequencing was performed in 11 of the 12 patients. A custom panel was designed to sequence the 169 genes known to be related to inherited muscular disease. First, a DNA library was prepared and amplified from genomic DNA. Then the coding exons and partial introns of 169 genes were selected by the SureSelect Target Enrichment System Kit (Agilent Technologies, Santa Clara, CA, USA). Sequencing was carried out on the GAIIX from Illumina (Santiago, CA, USA). Base calling was performed with the CASAVA 1.8 software (Illumina). Clean paired-end reads were aligned with the human genome reference sequence (UCSC hg19) using ANNOVAR. Insertion/deletions (indels) and single-nucleotide polymorphisms were also called with ANNOVAR. High VarQuality single-nucleotide polymorphisms were annotated with Perlscript into functional categories, such as nonsense, splice sites and missense. Homozygous variants and heterozygous variants appearing twice or more in 50 normal controls were excluded. Prediction of pathogenicity for novel missense variants was performed using Polyphen2.2.2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/) and Mutation Taster (http://www.mutationtaster.org). After analyses of the results from next-generation sequencing, candidate variants were verified using Sanger sequencing in patients and their parents.
Haplotype analysis
Haplotype analysis was performed in two unrelated probands with homozygous c.545A>G and their parents as well as seven probands with heterozygous c.545A>G to detect FKRP single-nucleotide polymorphisms. All were screened by direct sequencing for the following known polymorphisms: c.-34C>T; c.-84C>G; c.192C>T; c.1440C>T; c.1177G>C; and c.249C>T.
Results
Phenotypic features of patients
The patients were divided into two groups according to clinical manifestation.
Group 1: Patients with MDC1C phenotype (Patients 1–3)
Patients 1–3 were aged between 2 years and 3 years 3 months. All three children exhibited symptoms in the first week of life and presented motor development retardation, muscle weakness, hypotonia and tendon reflex not reduced. Two children (Patients 1 and 3) exhibited multi-joint contractures (elbow, hip, knee and ankle) and facial muscle weakness. The serum CK level of all three patients was highly elevated, ranging from 4437 to 7761 IU l−1. None of the three children exhibited gastrocnemius muscle hypertrophy. The maximum motor ability of these three patients was sitting unsupported with inability to walk. Patients 1 and 2 had normal to mild retarded mental development with nothing abnormal found in brain MRI. Patients 1 and 2 were clinically diagnosed with congenital muscular dystrophy at the first visit. Patient 3 presented with mental development retardation without eye involvement. Her MRI showed white matter abnormalities in the center of the cerebral hemisphere and the periventricular area and showed an enlarged frontal extracerebral space but not brainstem or cerebellar dysplasia or cerebellar cysts (Figure 1). According to these characteristics, Patient 3 was diagnosed with congenital muscular dystrophy type 1A (MDC1A) at the first visit (7 months).

Cranial brain MRI scan of Patient 3. The brain MRI of Patient 3 was taken when she was 5 months old. (a) White matter abnormalities are observed in the center of the cerebral hemisphere and the periventricular area. Signal increased in T2W and fluid attenuated inversion recovery (FLAIR). (b) No cerebellar cysts were observed, which is different from the MDC1C patients previously reported. No structural abnormality was found. Corpus callosum, internal capsule, basal ganglia and thalami were normal.
Group 2: Patients with LGMD2I phenotype (Patients 4–12)
Nine patients with LGMD2I were aged between 3 years 11 months and 42 years. Disease onset ranged from 1 year 5 months to 17 years of age. Their symptoms of onset were also different. Patient 4 presented with abnormal gait, Patients 5–7 and 9 exhibited frequent falling when walking, Patient 8 presented muscle weakness after fever, while Patients 10–12 exhibited nothing abnormal in motor ability. Their primary motor milestones and mental development were all normal. CK levels were moderately to significantly increased, ranging from 1451 to 12017 IU l−1. No patients presented facial muscle weakness or had become wheelchair bound. Patients 4–9 exhibited limb-girdle weakness, gastrocnemius muscle hypertrophy and Gowers’ sign. With ankle joint contractures and tendon reflex absence, Patients 4–7 fell down frequently, could not run, had difficulties in jumping, going up and down stairs and standing up from a squat. Patients 8–9 could run and go up and down stairs relatively easily. Patients 5, 7, 9 and 10 had cardiac involvement shown by electrocardiography (ECG) or ultrasound cardiography (UCG). The ECG of Patient 5 indicated complete right bundle branch block; the UCG of Patient 7 showed trivial aortic regurgitation and trivial tricuspid valve regurgitation; the UCG of Patient 9 showed a mildly enlarged left ventricle; Patient 10 showed indications of sinus arrhythmia and left ventricular high voltage by ECG and mild tricuspid and mitral regurgitation by UCG. Patients 5, 6, 7 and 9 had a pulmonary function test performed. Patient 5, who was 20 years 9 months old, showed a mild decrease in lung function; Patient 6, who was 25 years old, had mild mixed dysfunction of ventilation without subjective symptoms; 42-year-old Patient 7 presented with dyspnea; the test result of 5-year-old Patient 9 was normal. Based on the gender and clinical manifestations of patients, at the first visit, four male patients were diagnosed with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy, while others were diagnosed with LGMD, progressive muscular dystrophy, hyperCKemia and myocarditis.
Muscle pathology
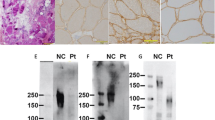
Muscle biopsy was performed on three MDC1C patients. All the three biopsy specimens had dystrophic changes, observed as increased variations in fiber size and proliferation of connective tissue seen in histochemical staining of tissue sections. In biopsy specimens of three patients, immunolabeling for α-DG with IIH-6 showed reduced glycosylated α-DG staining. In Patients 2–3, immunolabeling for laminin α2 with mAb 1922 revealed mildly reduced expression (Figure 2).

Skeletal muscle biopsy pathological analysis. (a, e, j, i, m) normal control; (b, f) Patient 1; (c, g, k) Patient 2; (d, h, l) Patient 3. (a–d) Hematoxylin–eosin staining (HE); (e–g) α-DG immunohistochemistry staining; (h, i) α-DG immunofluorescence staining; (j, k) laminin α2 immunohistochemistry staining; (l, m) laminin α2 immunofluorescence staining. The HE staining of Patients 1–3 (b–d) showed increased variations in fiber size and proliferation of connective tissue. The small fibers were round or polygonal. Immunolabeling for α-DG with IIH-6 of Patients 1–3 (f–h) showed reduced glycosylated α-DG staining. Immunolabeling for laminin α2 with mAb 1922 of Patients 2–3 (k, l) showed reduced expression. A full color version of this figure is available at the Journal of Human Genetics journal online.
Genotype features of patients
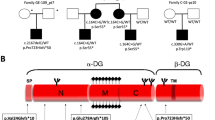
Direct sequencing of PCR-amplified genomic DNA and next-generation sequencing detected 15 mutations, including c.545A>G, c.948delC, c.350C>G, c.502T>C, c.1360T>A, c.938G>A, c.272delT, c.1067T>C, c.859_869delTTCGGCTGCAA, c.1391A>T, c.151G>T, c.204-206delCTC, c.941C>T, c.1248C>G and c.160C>G. The diagnosis and mutation of each patient are listed in Table 2. Thirteen of the 15 mutations are novel, except only c.545A>G and c.948delC.4 None of the novel FKRP mutations were found in 100 control population (or in the 1000 Genomes database (http://browser.1000genomes.org), Exome Aggregation Consortium database (http://exac.broadinstitute.org)). A schematic showing the mutations of the 12 patients on the FKRP protein sequence is shown in Figure 3. Interestingly, eight of the nine LGMD2I patients carried the c.545A>G mutation, which was homozygous in two patients and heterozygous in the other six patients. Haplotype analysis revealed that all patients carrying this mutation had the same haplotype (Table 3).

Schematic of the fukutin-related protein, indicating mutations. Mutations from three MDC1C patients and nine LGMD2I patients are indicated on the fukutin-related protein, where the cytoplasmic domain (amino acid 1–6), Golgi transmembrane domain (amino acid 7–29) and Golgi lumenal domain (amino acid 30–495) are indicated. The Golgi lumenal domain contains a stem region and catalytic domain. TMD=transmembrane domain. A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
Our study reported 12 Chinese patients with the dystroglycanopathies MDC1C and LGMD2I, carrying FKRP mutations. This is the first report of MDC1C patients in China with the unusual MRI manifestation observed here, which expands the clinical manifestation spectrum of MDC1C. Additionally, our study diagnosed nine LGMD2I patients, whereas previously only two mainland Chinese patients had been reported.5 Eight LGMD2I patients had the same c.545A>G mutation, indicating that LGMD2I is not rare in China and also that a founder mutation is present in this gene.
MDC1C is also called congenital muscular dystrophy-dystroglycanopathy with or without mental retardation type B5 (MDDGB5) (OMIM#606612). Previously reported clinical features of MDC1C are: onset in the first weeks of life; inability to walk; muscle hypertrophy; and highly elevated serum CK levels.7 These clinical features are observed in the patients described here. However, there is a difference compared with previous reports of the brain features as observed by MRI. The spectrum of brain changes in patients with MDC1C reported before include progressive brain atrophy, transient white matter changes, cerebellar cysts with or without structural brain changes and the structural brain involvement ranging from focal unilateral periventricular nodular heterotopia to marked cerebellar dysplasia and pontine hypoplasia.8, 9, 10, 11 The MRI of Patient 3 showed white matter abnormalities in the central region of the cerebral hemisphere and the periventricular area, which is similar to the changes observed in MDCIA patients. This case draws attention to the need for clinical physicians to make note of the possibility for a differential diagnosis and the importance of genetic diagnosis and long-term follow-up.
LGMD2I is also called limb-girdle muscular dystrophy-dystroglycanopathy type C5 (MDDGC5) (OMIM#606612) and is characterized by wide clinical variability, childhood to adult onset and a relatively benign course.2, 12, 13, 14 The nine LGMD2I patients reported here can be divided into three groups according to their clinical features, including mild late-onset LGMD phenotypic (Patients 4–7), DMD-like (Patients 8–9) and clinically asymptomatic as Patients 10–12. Dilated cardiomyopathy is a common feature of LGMD2I patients,7 but in our report three in nine patients had alternative types of cardiac involvement, rather than dilated cardiomyopathy. Because of the cardiac involvement, elevated myocardial enzyme spectrum and asymptomatic motor system, Patient 10 was diagnosed with myocarditis at their first visit, emphasizing the need for clinicians to distinguish these diseases. Another common symptom of LGMD2I is myalgia,15 but here it was only observed in Patient 7, who described the pain as a burning sensation. We hypothesized that the traction of nerves by connective tissue around dystrophic muscles could result in myalgia. In addition, respiratory involvement was observed in Patients 6–7 who were still ambulatory, suggesting that LGMD2I patients could develop respiratory failure in later stages of the disease, but no absolute correlation between skeletal muscle weakness and respiratory insufficiency has been shown.16 However, this suggests that routine monitoring of pulmonary function and early management of symptoms is important. As some LGMD2I patients are DMD-like, five male patients had been diagnosed with DMD at their first visit. Multiplex ligation-dependent probe amplification analysis and sequencing of the DMD gene were performed, with the results all negative. Finally, they were genetically diagnosed with LGMD2I after the identification of FKRP mutations by next-generation sequencing.
FKRP is composed of three noncoding exons and one exon encompassing the entire open reading frame, encoding FKRP (OMIM606596). A model for FKRP has been proposed, where FKRP resides in the Golgi cisternae of skeletal muscle fibers and forms disulfide-linked homodimers via an N-terminal interaction, consistent with that of a Golgi resident type II transmembrane protein.17 FKRP is ribitol 5-phosphate transferase critical for the appropriate glycosylation of α-DG,18 a peripheral membrane protein expressed in several tissues such as muscles and brains.19 Glycosylated α-DG links the subsarcolemmal actin-associated cytoskeleton via β-dystroglycan and dystrophin, and it also links the extracellular matrix via laminin α2.20 Mutations in FKRP reduce the glycosylation of α-DG, which disrupts these interactions, finally resulting in defects of basement membranes in both skeletal muscle and brain.21, 22 Our findings in muscle pathology, including reduced glycosylated α-DG staining and reduced expression of laminin α2 in the patient biopsies, provide further evidence of an association between α-DG and laminin α2.1 Effects on the brain were limited to Patients 2–3, who exhibited mental retardation, and Patient 3 with white matter abnormalities, while other patients exhibited normal mental development and brain MRI. Variability between the patients may be a result of different α-DG glycosylation levels.
The level of α-DG glycosylation depends on the severity of the effect of the particular FKRP mutation on protein function.3 The FKRP mutations in three of the MDC1C patients were compound heterozygous mutations. Compared with Patient 1 with missense mutations, Patient 2 had a nonsense mutation and Patient 3 had a frameshift mutation, which may have a more severe effect on FKRP function, giving rise to mental retardation and brain involvement. Eight of the nine LGMD2I patients in our cohort carried the c.545A>G mutation, and three of the six LGMD2I patients previously reported in Taiwan also had the same mutation.4 According to haplotype analysis, the eight patients with the c.545A>G mutation had the same haplotype, indicating that it may be a founder mutation. The c.948delC mutation, a common mutation in LGMD2I patients of Taiwan,4 was also detected in Patient 6 in our study. However, the Chinese founder c.545A>G is different from the founder c.826C>A (p.Leu276Ile) mutation observed in Europe.23 Despite the difference in the mutation, a similarity exists between these two groups of patients in that they present milder clinical manifestations than patients with other FKRP mutations. Moreover, Patients 11–12 who are homozygous for the c.545A>G mutation have not exhibited dystrophic phenotypes to date, and their phenotypes are less severe than patients with compound heterozygous c.545A>G mutations. This is also consistent with the manifestation of the c.826C>A founder mutation in European patients, although follow-up of Patients 11–12 is necessary as they are still in childhood. The homozygous c.826C>A mutation patients had later onset, milder clinical progression and less muscle weakness compared with compound heterozygous patients with the c.826C>A mutation.24 The mild progression in patients with LGMD2I who are homozygous for the c.826C>A mutation resembles that of the homozygous FKRP c.826C>A mouse model.25 These patients’ relatively mild manifestation was presumably owing to the less severe effect c.545A>G and c.826C>A had on FKRP protein.
To date, there is no effective treatment available for MDC1C or LGMD2I, although adeno-associated virus serotype 9 vector-mediated gene therapy has been shown to be highly effective in the FKRP mutant mouse model with a missense mutation, P448L. The gene therapy resulted in systemic FKRP expression in all striated muscles examined and consequently restored functional glycosylation of α-DG in the skeletal and cardiac muscles.26 In addition, the potential benefit of corticosteroid treatment in patients with LGMD2I has been previously reported in two patients with subacute onset triggered by viral infection.27 Recently, a case of subacute necrotizing myopathy with the homozygous FKRP mutation L276I was reported to respond well to immune therapies, highlighting the potential benefit of immunomodulatory therapies in LGMD2I patients with subacute onset.28 The LGMD2I patients described in this study were recommended to undertake moderate-intensity endurance training as a safe and effective method to increase exercise performance and daily function, emphasizing the importance of rehabilitation training.29
Our study provided clinical and genetic diagnosis for MDC1C and LGMD2I patients in China. The founder mutation c.545A>G found in LGMD2I patients is different from the one in European patients, which indicates the ethnic difference in these gene mutations. Next-generation sequencing had an important role in the genetic diagnosis of these patients, with 13 novel mutations in FKRP significantly expanding the mutation spectrum of MDC1C and LGMD2I. This will improve the early diagnosis of these diseases.
References
Brockington, M., Blake, D. J., Prandini, P., Brown, S. C., Torelli, S., Benson, M. A. et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 69, 1198–1209 (2001).
Brockington, M., Yuva, Y., Prandini, P., Brown, S. C., Torelli, S., Benson, M. A. et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 10, 2851–2859 (2001).
Beltran-Valero de Bernabe, D. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 41, e61–e61 (2004).
Liang, W. C., Hayashi, Y. K., Ogawa, M., Wang, C. H., Huang, W. T., Nishino, I. et al. Limb-girdle muscular dystrophy type 2I is not rare in Taiwan. Neuromuscul. Disord. 23, 675–681 (2013).
Hong, D., Zhang, W., Wang, W., Wang, Z. & Yuan, Y. Asian patients with limb girdle muscular dystrophy 2I (LGMD2I). J. Clin. Neurosci. 18, 494–499 (2011).
Kang, P. B., Feener, C. A., Estrella, E., Thorne, M., White, A. J., Darras, B. T. et al. LGMD2I in a North American population. BMC Musculoskelet. Disord. 8, 115 (2007).
Mercuri, E., Brockington, M., Straub, V., Quijano-Roy, S., Yuva, Y., Herrmann, R. et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann. Neurol. 53, 537–542 (2003).
Topaloglu, H., Brockington, M., Yuva, Y., Talim, B., Haliloglu, G., Blake, D. et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 60, 988–992 (2003).
Mercuri, E., Topaloglu, H., Brockington, M., Berardinelli, A., Pichiecchio, A., Santorelli, F. et al. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch. Neurol. 63, 251–257 (2006).
Louhichi, N., Triki, C., Quijano-Roy, S., Richard, P., Makri, S., Meziou, M. et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics 5, 27–34 (2004).
Quijano-Roy, S., Marti-Carrera, I., Makri, S., Mayer, M., Maugenre, S., Richard, P. et al. Brain MRI abnormalities in muscular dystrophy due to FKRP mutations. Brain Dev. 28, 232–242 (2006).
Poppe, M., Cree, L., Bourke, J., Eagle, M., Anderson, L. V., Birchall, D. et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 60, 1246–1251 (2003).
de Paula, F., Vieira, N., Starling, A., Yamamoto, L. U., Lima, B., de Cassia Pavanello, R. et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: the other end of the spectrum. Eur. J. Hum. Genet. 11, 923–930 (2003).
Schottlaender, L. V., Petzold, A., Wood, N. & Houlden, H. Diagnostic clues and manifesting carriers in fukutin-related protein (FKRP) limb-girdle muscular dystrophy. J. Neurol. Sci. 348, 266–268 (2015).
Mathews, K. D., Stephan, C. M., Laubenthal, K., Winder, T. L., Michele, D. E., Moore, S. A. et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology 76, 194–195 (2011).
Poppe, M., Bourke, J., Eagle, M., Frosk, P., Wrogemann, K., Greenberg, C. et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann. Neurol. 56, 738–741 (2004).
Alhamidi, M., Kjeldsen Buvang, E., Fagerheim, T., Brox, V., Lindal, S., Van Ghelue, M. et al. Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibres and forms disulfide-linked homodimers via an N-terminal interaction. PLoS ONE 6, e22968 (2011).
Kanagawa, M., Kobayashi, K., Tajiri, M., Manya, H., Kuga, A., Yamaguchi, Y. et al. Identification of a post-translational modification with ribitol-phosphate and its defect in muscular dystrophy. Cell Rep. 14, 2209–2223 (2016).
Henry, M. D. & Campbell, K. P. Dystroglycan inside and out. Curr. Opin. Cell Biol. 11, 602–607 (1999).
Ibraghimov-Beskrovnaya, O., Ervasti, J. M., Leveille, C. J., Slaughter, C. A., Sernett, S. W. & Campbell, K. P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702 (1992).
Michele, D. E., Barresi, R., Kanagawa, M., Saito, F., Cohn, R. D., Satz, J. S. et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418, 417–422 (2002).
Montanaro, F. & Carbonetto, S. Targeting dystroglycan in the brain. Neuron 37, 193–196 (2003).
Frosk, P., Greenberg, C. R., Tennese, A. A., Lamont, R., Nylen, E., Hirst, C. et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum. Mutat. 25, 38–44 (2005).
Sveen, M. L., Schwartz, M. & Vissing, J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann. Neurol. 59, 808–815 (2006).
Krag, T. O. & Vissing, J. A new mouse model of limb-girdle muscular dystrophy type 2I homozygous for the common L276I mutation mimicking the mild phenotype in humans. J. Neuropathol. Exp. Neurol. 74, 1137–1146 (2015).
Xu, L., Lu, P. J., Wang, C. H., Keramaris, E., Qiao, C., Xiao, B. et al. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of alpha-dystroglycan and improves muscle functions. Mol. Ther. 21, 1832–1840 (2013).
Darin, N., Kroksmark, A. K., Ahlander, A. C., Moslemi, A. R., Oldfors, A. & Tulinius, M. Inflammation and response to steroid treatment in limb-girdle muscular dystrophy 2I. Eur. J. Paediatr Neurol. 11, 353–357 (2007).
Svahn, J., Streichenberger, N., Benveniste, O., Menassa, R., Michel, L., Fayolle, H. et al. Significant response to immune therapies in a case of subacute necrotizing myopathy and FKRP mutations. Neuromuscul. Disord. 25, 865–868 (2015).
Sveen, M. L., Jeppesen, T. D., Hauerslev, S., Krag, T. O. & Vissing, J. Endurance training: an effective and safe treatment for patients with LGMD2I. Neurology 68, 59–61 (2007).
Acknowledgements
We thank the doctors, patients and their families for their participation in this study, and thank the native-English-speaking scientific editors from Edanz Group for the editing of our manuscript. This work was supported by grants from the National Natural Science Foundation of China (81271400) (81571220) and the National Basic Research Program of China (2012CB944602).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Fu, X., Yang, H., Wei, C. et al. FKRP mutations, including a founder mutation, cause phenotype variability in Chinese patients with dystroglycanopathies. J Hum Genet 61, 1013–1020 (2016). https://doi.org/10.1038/jhg.2016.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.94
This article is cited by
-
Clinical, pathological, imaging, and genetic characterization in a Taiwanese cohort with limb-girdle muscular dystrophy
Orphanet Journal of Rare Diseases (2020)
-
The clinical spectrum and genetic variability of limb-girdle muscular dystrophy in a cohort of Chinese patients
Orphanet Journal of Rare Diseases (2018)
