
Abstract
Dystrophinopathy is a group of inherited diseases caused by mutations in the DMD gene. Within the dystrophinopathy spectrum, Duchenne and Becker muscular dystrophies are common X-linked recessive disorders that mainly feature striated muscle necrosis. We combined multiplex ligation-dependent probe amplification with Sanger sequencing to detect large deletions/duplications and point mutations in the DMD gene in 613 Chinese patients. A total of 571 (93.1%) patients were diagnosed, including 428 (69.8%) with large deletions/duplications and 143 (23.3%) with point mutations. Deletion/duplication breakpoints gathered mostly in introns 44–55. Reading frame rules could explain 88.6% of deletion mutations. We identified seventy novel point mutations that had not been previously reported. Spectrum expansion and genotype–phenotype analysis of DMD mutations on such a large sample size in Han Chinese population would provide new insights into the pathogenic mechanism underlying dystrophinopathies.
Similar content being viewed by others

Introduction
The dystrophin gene (DMD, MIM no. 300377), located at Xp21.2, is one of the largest known human genes, covering 2.2 Mb and containing 79 exons. More precisely, it is located in the region from base pair 31 119 221 to base pair 33 339 608 on the X chromosome. DMD is highly complex, containing at least eight independent, tissue-specific promoters and two polyA-addition sites. Furthermore, dystrophin RNA is differentially spliced, producing a range of different transcripts encoding a large set of protein isoforms. The DMD gene encodes a protein called dystrophin (encoded by the Dp427 transcript), which is a large, rod-like cytoskeletal protein. This protein is primarily located in skeletal muscles (used for movement), and cardiac muscle. Small amounts of dystrophin are present in nerve cells in the brain.
Dystrophinopathy is a group of inherited muscle diseases caused by defects in the dystrophin protein. Severe end of the dystrophinopathy spectrum includes progressive muscle diseases that are classified as Duchenne/Becker muscular dystrophy (DMD/BMD) when skeletal muscle is primarily affected and as DMD-associated dilated cardiomyopathy (DCM) when the heart is primarily affected.1
DMD and BMD are common X-linked recessive disorders that are caused by pathogenic mutations in DMD. DMD is the most frequent but lethal inherited muscle disease in children, affecting 1 in 3500 live-born males,2 whereas BMD is a milder allelic form with a reported incidence of 1 in 30 000 live-born males.3 DMD, the severe form, is characterized by progressive skeletal muscle necrosis, pseudo-hypertrophy in the calf muscle and Gowers’ sign. DMD patients are usually first recognized before 5 years of age, lose independent walking ability by the age of 12 and die from cardiac or respiratory failure at around 20 years of age.4, 5 BMD, with its milder manifestation, often progresses slowly and some patients can live up to 60 years of age.6
More than 2000 pathogenic variants in the DMD gene have been identified in people with the Duchenne and Becker forms of muscular dystrophy. According to the previous research reports,7, 8 ~60–65% of DMD or BMD cases are due to deletions of one or more exons, 5–10% due to duplications of one or more exons and the rest are attributed to factors such as point mutations. In this study, we implemented a combination of multiplex ligation-dependent probe amplification (MLPA) with Sanger sequencing to comprehensively detect large fragment deletion or duplication mutations and point mutations in the DMD gene in 613 Chinese patients who showed clinical phenotype compatible with DMD/BMD. We analyzed DMD gene deletion/duplication mutations and point mutations and provided a more comprehensive description of DMD gene mutations in Han Chinese population.
Materials and methods
Patients
A group of 613 unrelated male probands from 613 cases referred to the State Key Laboratory of Medical Genetics were studied. All patients were diagnosed on the basis of pre-determined inclusion criteria that included either (a) clinical symptoms, referring to serum CK levels, age of onset, age at loss of ambulation, calf muscle hypertrophy, Gower’s sign, presence of cardiomyopathy and electromyographic patterns, suggestive for DMD or BMD and an X-linked family history; or (b) muscular biopsy revealing abnormity in dystrophin expression by immunofluorescence, immunohistochemistry or immunoblot. 596 patients were severely affected, diagnosed as DMD, 17 were considered to represent a milder form (BMD). Fifty females, immediately related to the probands, without any clinical dystrophinopathy phenotype were voluntary to be involved in our research. Our genetic research was approved by the institutional review board of Central South University, and had informed consent from the patients or their legal guardians.
Genomic DNA extraction
After family members of patients voluntarily signed the informed consent form, 3–4 ml peripheral venous blood (EDTA anticoagulant) was collected from the patients. Genomic DNA was extracted using the standard phenol-chloroform method.
MLPA
The SALSA MLPA P034/P035 (MRC Holland, Amsterdam, The Netherlands) kit was used in accordance with the manufacturer’s instructions. The MLPA samples consisted of approximately 200 ng of genomic DNA. Denaturation, hybridization, ligation and amplification were carried out using the ABI 2720 PCR amplification. The PCR reaction conditions included 33 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 60 s each, followed by a final extension at 72 °C for 20 min to allow adequate probe hybridization with SALSA probe mix P034 (DMD gene exons 1–10, 21–30, 41–50 and 61–70) and P035 (DMD gene exons 11–20, 31–40, 51–60 and 71–79). Amplification products were analyzed using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Forster City, CA, USA). The data obtained were analyzed using Coffalyser 9.4 (MRC Holland). For the samples with a suspected single exon deletion, PCR and direct sequencing were applied for further verification.
Structural analysis of DMD gene introns
To explore the molecular mechanism of DMD gene breaking in hotspots, all 78 introns of the DMD gene were analyzed using Repeatmasker 19.0 (http://www.repeatmasker.org/). The sequence data of the introns (NM_004006.2) was obtained from the UCSC website.
Reading frame analysis
The reading frame of large fragment deletion mutations was analyzed using the online DMD exonic deletions/duplications reading-frame checker 1.9 (updated 2009, http://www.humgen.nl/scripts/DMD_frame.php).
Sanger sequencing
PCR amplification and direct DNA sequencing were performed for cases in which MLPA found no exon deletion/duplication. A total of 86 pairs of forward and reverse primers were designed,9 which were complementary to all 79 exons, the 5′ promoter region, 3′ downstream region and exon–intron junctions (Supplementary Data 1). The amplified products were purified and sequenced on an automated DNA Sequencer (Model 3130; Applied Biosystems). Sequencing results were interpreted using Lasergene 7.0.
Analysis of DMD mRNA
Dystrophin encoded by DMD is primarily expressed in skeletal and cardiac muscles. By querying the GeneCards, transcription of DMD in peripheral leukocytes have been recorded. Because of unavailability in muscle sampling, we collected fresh peripheral blood from patients to carry out mRNA analysis; only a small part had taken mRNA analysis. Within 6 h of collecting peripheral venous blood, as described above, RNA was extracted using the standard TRIzol method then changed to DNA by reverse transcription using the RevertAid First Strand cDNA Synthesis Kit (Thermo, Waltham, MA, USA) according to the manufacturer’s instructions. Extracted cDNA was stored at −20 °C. Special primers designed for different splice-site mutations could give verification for functional changes of DMD gene. At the same time, we set one normal control and one positive control which showed the testing sample abnormal indeed. Furthermore, analysis of DMD mRNA provided a chance to find mutations in deep introns.
Results
This study combined MLPA with Sanger sequencing to detect DMD gene mutations in 613 probands with DMD or BMD. Large fragment deletion or duplication mutations were found in 428 (69.8%) patients, and 143 (23.3%) patients were found to have point mutations (Table 1). No mutation in the DMD gene was detected by either method in 42 (6.9%) patients.
Large fragment deletion/duplication mutations
Of the 613 probands, in 369 (60.2%), large fragment deletions encompassing one or more exons were observed, of which 363 were in DMD patients and 6 in BMD patients (Supplementary Data 2). Large deletions occurred most frequently in the central region of DMD gene exons 45–54 and near the 5′ end region in exons 3–22, accounting for 71.8% (265/369) and 18.4% (68/369) of deletions, respectively (Figure 1). We categorized large deletions found in our research into 132 patterns. Single exon deletion mutation, which was observed in 95 out of 369 patients, was the most common pattern. The largest fraction of all 132 deletion patterns comprised exon 45 deletions (6.2%) and exon 48–50 deletions (6.2%).

Distribution of 369 large fragment deletions and 62 large fragment duplications. Blue bars show distribution of 369 deletions in 132 patterns. Numbers below bars stand for the cumulative frequencies of each different pattern of deletion. Red bars show distribution of 62 duplications in 49 patterns. Numbers below bars stand for the cumulative frequencies of each different pattern of duplication (see details of 428 cases with deletions/duplications in Supplementary Data 2).
Deletions of exon 45, exons 45–47 and exons 45–55 were found in both DMD and BMD patients. In particular, the largest fragment deletion, spanning exons 1–63, was identified in a suspected DMD patient with impaired hearing who had typical clinical symptoms of DMD. Scanning genomic DNA using the Human CytoSNP-12 beads array (Illumina, San Diego, CA, USA), we found a 5 Mb deletion at chromosome X in this patient.
Of all 613 cases, 62 large fragment duplications, classified into 49 patterns, were detected in 59 (9.6%) patients (Supplementary Data 2). Single exon duplication, which was the most frequent type, occurred in 14 (22.6%) cases. MLPA results indicated that exons 3–9 were the most commonly duplicated regions (Figure 1). Three complex gene rearrangements involving two duplicated regions were reported: dup 1 and dup 3–16; dup 1 and dup 45–49; and dup 24–37 and dup 61–64. These three patients were diagnosed with typical DMD according to clinical symptoms.
Single exon deletion or duplication is most frequently detected in our study. All were verified by PCR using special primers for different exon and direct sequencing. Results of 95 single exon deletions and 14 single exon duplications were final reports according to both of MLPA and Sanger sequencing.
Reading frame
There was no direct association between the size of DMD gene deletion and disease severity. However, this depended on whether the large fragment deletion mutation changed the DMD gene reading frame or not. According to the reading frame rules first reported by Monaco et al.,10 an in-frame deletion, which does not change the reading frame, usually leads to milder BMD, whereas out-of-frame deletions that break the original DMD gene reading frame cause more severe DMD. DMD gene with an out-of-frame deletion mutation generates abnormal RNA transcription and produces a truncated protein that degrades easily.
In our study, 369 patients with large deletions, including 363 DMD and 6 BMD, were analyzed by using online DMD exonic deletions/duplications reading-frame checker 1.9. The data indicated that reading frame rules could interpret the relationship between large deletions and clinical phenotype in 327 (88.6%) cases. Nevertheless, 41 DMD patients with in-frame deletions and 1 BMD patient with an out-of-frame deletion in the DMD gene did not follow the reading frame rules (Supplementary Data 3).
Large deletion and duplication breakpoint distribution
Large deletion breakpoints gathered mostly in the introns 44–55, where the DMD gene-center deletion hotspots were situated (Figure 2). However, near the 5′-end region, where deletions occurred second most frequently, breakpoints were dispersed. Of all 738 deletion breakpoints, 84 (11.4%) were located in intron 44 which was most frequently involved. Those located in the intron 44 tended to be 5′ starting breakpoints of deletions and 78 breakpoints located in the intron 51 tended to be 3′ ending ones of deletions. Near the 5′ end of the DMD gene, there were more breakpoints located in intron 2 (21 breakpoints) and intron 7 (26 breakpoints) than other introns, nearly all of which were 5′-starting breakpoints of deletions (19/21 and 18/26, respectively). Compared with deletion breakpoints, duplication breakpoints had a more disperse distribution (Figure 2). Intron 2 was most frequently involved and accounted for 12.1% (15/124).

Distribution of large-deletion and duplication breakpoints. (a) Distribution of 738 breakpoints in 369 DMD gene gross deletions. (b) Distribution of 124 breakpoints in 62 DMD gene gross duplications.
Structural analysis of introns
Previous research showed that DMD gene deletion or duplication breakpoints almost all located in introns.11 However researchers also reported few DMD gene deletion breakpoints distributed on exons.12 Analysis of all 78 introns of the DMD gene using Repeatmasker 19.0 revealed that the proportion of interspersed repetitive sequences in each intron averaged 28.27% (Supplementary Data 4). These repeats are important in introns, especially long interspersed nuclear elements (LINEs), which form the main sequence of DMD introns.
862 breakpoints in 428 patients with deletion/duplication were observed in all introns except introns 10, 24, 35–36, 38, 58, 65–66, 68–73 and 75–78. Using canonical correlation analysis (SPSS 19.0, Armonk, NY, USA) to calculate the related coefficient, Rs=0.383 (P⩽0.05), the result demonstrated that the distribution of DMD gene breakpoints showed a significant positive correlation with the proportion of interspersed repetitive sequences in each intron.
In our research, introns 44, 45 and 50 were the most gathering regions, being involved in 107, 80 and 98 cases, respectively. The proportions of interspersed repetitive sequences for these introns were 30.36, 53.64 and 45.62%, which were all above the average value of 28.27% (Figure 3).

The distribution of DMD gene breakpoints and the proportion of interspersed repetitive sequences in each intron (introns 44–55).
Sanger sequencing results
The entire coding sequence of the DMD gene was sequenced for patients who had tested negative by MLPA. Direct Sanger sequencing identified 143 mutations in 185 unrelated probands with negative MLPA, which accounted for 23.3% of all 613 cases. Sixty-four nonsense mutations were identified in 70 cases (11.4%). Mutations disrupting the splice-site consensus sequences were detected in 21 cases (3.4%). Small deletions/insertions (one to several hundred nucleotides) were identified in 52 cases (8.5%). These three patterns of mutations were dispersed across the entire DMD gene and no clustering was identified.
Nonsense mutation was the second most common mutation detected in our study. Of 70 cases with nonsense mutations identified, 42 (60%) were cytosine (C) to thymine (T) and 20 (28.6%) were not recorded in the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/) or LOVD database (Table 2). When a nonsense mutation occurs in the DMD gene, the codon for an amino acid changes into a termination codon, disrupting peptide synthesis and producing a truncated protein. All of these nonsense mutations led to clinical DMD.
We observed 52 small deletions and insertions, ranging from 1 to 22 bp, and 36 of them were new reports according to HGMD database and Leiden Open Variation Database (LOVD, http://www.lovd.nl/3.0/home) (Table 3). In addition, 32 small deletions, 17 small insertions and 3 small deletions with insertions were observed in this study. Small deletions or insertions usually changed the DMD reading frame, making a termination codon appear early and translating a truncated protein. Thus, small deletions/insertions generally produced clinical DMD.
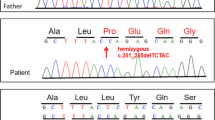
Splice site mutations mean nucleotide changes disrupting the splice-site consensus sequences. In 21 cases, splice-site mutations were determined to be the disease causing mutations (Table 4). The DMD gene with a splice-site mutation transcribes abnormal RNA, leading to codons becoming lost or changed. In 20 out of 21 cases, the nucleotide changes ranged within 1–2 bases around the 3′-terminal of exons. Analysis of mRNA was carried out only in one case with mutation c.531-16T>G, which was not consistent with this. Analysis of DMD mRNA identified that c.531-16T>G did indeed produce an mRNA variant with an excess length of 15 bp (Figure 4). Seven splice-site mutations were found in HGMD database and LOVD database, which stated clearly whether or not they were pathogenic. Other 13 splice-site mutations were not processed further because there was no chance to collect more peripheral venous blood from the patients. These 13 splice-site mutations were evaluated by the splice-site predictor NNSPLICE version 0.9 (January 1997) and all were predicted to have a disease-inducing potential.

Sanger sequencing result of one patients shows a T>G mutation at c.531-16 in intron6 of DMD gene. (a) Orange box show the previous normal splice-site and the later active one. (b) DMD gene with c.531-16T>G produces a 15 bp longer mRNA than normal.
In our study there were still 42 (6.9%) patients with negative MLPA for whom direct sequencing detected no mutations in the DMD gene. Many benign-like single nucleotide variants (SNVs) and small indels were observed. In mutation analysis, benign-like SNVs and small indels could be categorized into three classes: (1) SNPs recorded in dbSNP or LOVD; (2) SNVs or small indels, unrecorded in databases or happened within introns and predicted benign by NNSPLICE; and (3) SNVs, unrecorded in database or happened within exons, and predicted benign by SIFT and PolyPhen-2.
Family history and detection of carriers
Detailed genealogical information was available for all of the 613 apparently unrelated clinically diagnosed DMD or BMD patients. A family history of DMD or BMD was present in 114 cases, while 499 cases were sporadic. In the 499 probands without family history, 460 (92.1%) positive results containing large deletions/duplications and point mutations were detected, and 111 (97.4%) positive results were found in the 114 cases with a family history. The statistical X2-test was performed to analyze the sample rates of probands with and without family history, which were found to be 97.4 and 92.1%, respectively. Hence, there was no significant difference between them. In other words, positive detection rate had no correlation with family history of DMD or BMD probands. Of 50 females immediately related to the probands, who were subjected to a test to determine if they carried the DMD gene mutation, 35 were found to be positive while 15 were negative.
Clinical phenotypic variety and genotype
DMD usually appears before 5 years of age with high serum creatine kinase and an absence of dystrophin protein in muscle biopsy. The patients in our study had an average onset age of approximately 3 years. Three Hundred and forty-five patients had taken a serum CK test before being interviewed by the geneticists in our clinic. The CK value showed a large range from 234 to 50744 U l−1, which was higher than normal ones varied according to the age. And it was up to 11537 IU l−1 on average which was nearly 50–100 folds the normal value. DMD patients may be characterized by mild intellectual disability although a few patients present with severe intellectual disability. However, most DMD patients came to our lab for the DMD gene test because of high serum creatine kinase, without attempting the intelligence quotient test. 12 patients with detailed records of intelligence including six point mutations, five gross deletions and one gross duplication were identified.
Discussion
The human genome contains a large number of repetitive DNA sequences, including tandem repeats and interspersed repetitive sequences. The latter are generally moderately repetitive sequences that are divided into two categories according to the length of the repetitive units, short interspersed nuclear elements (SINEs, <500 bp) and LINEs (>1000 bp). Deletion or duplication breakpoints in the DMD gene had clear aggregation. The clusters of breakpoints indicated that there were some special structures leading to an unstable, easily broken gene in these regions. Instability of the human genome is caused primarily by homologous or nonhomologous recombination events, and repetitive sequences are important targets for homologous recombination.13 Our research revealed that the DMD gene contained 28.27% repeat elements on average in each intron. LINEs in particular constitute the key structures in DMD gene introns.
Through this research we discovered that the proportion of interspersed repetitive sequences in DMD gene breakpoint clusters, introns 44–55, was higher than the average value (28.27%). The proportion was lower than 15% in 12 of the 78 introns, and only 18 of the 862 breakpoints (2.1%) were found to be located in these regions. In contrast, there were 14 introns in which the proportion was higher than 45%, and 403 (46.8%) breakpoints were located here. According to canonical correlation analysis of the data, the distribution of DMD gene breakpoints correlates positively with the proportion of interspersed repetitive sequences in each intron, Rs=0.383 (P⩽0.05). To elaborate, the higher the proportion of interspersed repetitive sequences, the more possible breakpoints there are. However, some introns did not follow this rule. Introns 25 and 67 contained many repeat elements, 50.94 and 45.99%, respectively, but we identified only one breakpoint in either intron. The data reported here suggested that gene breaking was not directly caused by large percentage of SINEs and LINEs in DMD gene introns. However, interspersed repetitive sequences might be the molecular basis of DMD gene-breaking hotspot instability.
In 1988, Monaco et al.10 first proposed the frame-shift hypothesis, considering the severity of a phenotype to be related to changes in the open reading frame. According to these reading frame rules, mutations that change the reading frame and produce truncated proteins would lead to severe DMD; whereas mutations that do not alter the reading frame lead to mild BMD. Reading frame rules can explain most connections between DMD genotype and phenotype. In our research, 41 DMD patients and 1 BMD patient (11.4% overall 369 gross deletion patients) did not follow the reading frame rules, which is in line with other reports.14, 15 We discovered that deletions of exon 45, exons 45–47 and exons 45–55 could cause DMD or BMD in different individuals. Deletion of exon 45 was out-of-frame mutation while both deletion of exons 45–47 and deletion of exons 45–55 were in-frame mutations. Although the DMD gene with an in-frame mutation could be transcribed and translated, reaching the carboxyl terminus of the protein encoded, the incomplete protein produced might be nonfunctional because of substantial loss of genetic information. Alternatively, the in-frame mutations occurring in regions essential to dystrophin, such as the cysteine-rich region, produce unstable proteins. Furthermore, deletions or duplications may cause abnormal splicing in RNA, disturbing the reading frame. Recently, researchers have discovered a new phenomenon of partial deletion of exons in nine DMD samples, indicating that breakpoints might be located on exons.12 Each of these aforementioned views could probably explain the DMD in patients with in-frame mutations. DMD gene duplication mutations are usually but not always tandem duplications. Therefore, MLPA test results are unreliable predictors of the impact of changes in the reading frame.16
In the whole DMD gene, 143 point mutations including 70 (48.9%) nonsense mutations, 52 (36.4%) small deletions/insertions and 21 (14.7%) splice-site mutations were found without clusters. Nonsense mutations were caused mostly by C changing into T. To elaborate, C in CpG sites is easily methylated to form 5'-methyl-cytosine then deaminated to form T. Therefore, CpG sites might be DMD gene mutation hotspots.14, 17 The presence of nonsense mutation hotspots in CpG sites of the DMD gene remains controversial. Many reports of analysis of a large sample of DMD gene mutations mentioned different CpG sites as spots of frequent mutations.14, 15, 18 Our research has enriched the DMD gene mutation spectrum with 70 point mutations unrecorded in the database.
Intellectual disability to a mild degree is a pleiotropic effect of mutations in the DMD gene. Dystrophin mRNA has been found in the human brain, which supports this statement.19, 20 Past research has indicated that those with a severe mental defect had later age of onset and confinement to wheelchair and a less marked decrease in creatine kinase levels with age.21, 22 No significant intelligence quotient difference were found between patients with promoter deletions and those without, nor was any relationship between length of deletion and full scale intelligence quotient observed. However, patients with distal deletions are more likely to be mentally challenged than those with proximal deletions.18 Detailed records of intelligence were available for 12 patients in our research, who had five gross deletions which were located central region hotspots (exons 45–54). Although our sample size was small, experimental results were in line with the literature.23
At present, some studies about DMD gene mutations of Chinese patients with DMD or BMD have been published. Juan Yang et al.24 used MLPA to detect the mutations in 1053 Chinese patients with DMD/BMD, 59.35 and 11.21% of which were deletions and duplications, respectively. However they concentrated more on the deletion and duplication mutations and only performed Sanger sequencing in 20 patients with negative MLPA. Xiaoming Wei et al.12 introduced a new single-step method, which targeted the next-generation sequencing, for the genetic analysis of 89 DMD patients. Our research combined MLPA with Sanger sequencing to comprehensively detect large deletions/duplications and point mutations in the DMD gene in 613 Chinese patients with DMD or BMD allowed a total of 571 (93.1%) patients to be diagnosed clearly on a molecular level. MLPA identified 428 (69.8%) mutations comprising 369 deletions and 59 duplications. Because of its relatively low testing times and economical nature, MLPA should be the preferred method of testing for DMD and BMD. There remained 42 patients with suspected DMD or BMD for whom no mutation was found by either method. Collection of peripheral venous blood from the patients to extract RNA and carry out RT-PCR to establish whether or not the transcription is normal would help us to find mutations in deep introns and make further diagnosis for these patients.15
In conclusion, our research implemented a combination of MLPA with Sanger sequencing to make a genotype–phenotype analysis of DMD mutations on such a large sample, and provided a comprehensive interpretation of DMD gene mutation in the Han Chinese population. We had uncovered many new and previously unreported mutations, expanding the DMD gene mutation spectrum. It provided new insights into the pathogenic mechanism underlying dystrophinopathies and a molecular basis for study into the mechanism of DMD gene mutations and exploration of treatments for DMD.
References
Darras, B. T., Miller, D. T. & Urion, D. K. Dystrophinopathies. 2000 Sep 5 [Updated 2014 Nov 26]. Pagon, R. A., Adam, M. P. & Ardinger, H. H. GeneReviews® [Internet], (Seattle (WA): University of Washington, Seattle, Seattle, 1993-2015).
Emery, A. E. Population frequencies of inherited neuromuscular diseases: a world survey. Neuromuscul. Disord 1, 19–29 (1991).
Bushby, K. M., Thambyayah, M. & Gardner-Medwin, D. Prevalence and incidence of Becker muscular dystrophy. Lancet 337, 1022–1024 (1991).
Bushby, K., Finkel, R., Birnkrant, D. J., Case, L. E., Clemens, P. R., Cripe, L. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet. 9, 77–93 (2010).
Essex, C. & Roper, H. Lesson of the week: late diagnosis of Duchenne's muscular dystrophy presenting as global developmental delay. BMJ 323, 37–38 (2001).
Bushby, K. M., Gardner-Medwin, D., Nicholson, L. V., Johnson, M. A., Haggerty, I. D., Cleghorn, N. J. et al. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. II. Correlation of phenotype with genetic and protein abnormalities. J. Neurol. 240, 105–112 (1993).
Prior, T. W. & Bridgeman, S. J. Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J. Mol. Diagn. 7, 317–326 (2005).
Lalic, T., Vossen, R. H., Coffa, J., Schouten, J. P., Guc-Scekic, M., Radivojevic, D. et al. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet. 13, 1231–1234 (2005).
Li, S. Y., Sun, X. F., Li, Q., Zhang, H. M. & Wang, X. M. Association of mutation types and distribution characteristics of dystrophin gene with clinical symptoms in Chinese population. Yi Chuan 33, 251–254 (2011).
Monaco, A. P., Bertelson, C. J., Liechti-Gallati, S., Moser, H. & Kunkel, L. M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1988).
Blonden, L. A., Grootscholten, P. M., den Dunnen, J. T., Bakker, E., Abbs, S., Bobrow, M. et al. 242 breakpoints in the 200-kb deletion-prone P20 region of the DMD gene are widely spread. Genomics 10, 631–639 (1991).
Wei, X. M., Dai, Y., Yu, P., Qu, N., Lan, Z., Hong, X. et al. Targeted next-generation sequencing as a comprehensive test for patients with and female carriers of DMD/BMD: a multi-population diagnostic study. Eur. J. Hum. Genet. 22, 110–118 (2014).
Sheng, W. L., Chen, J. Y., Zhu, L. F. & Liu, Z. Relationship between repeat elements and instability and gene deletion of DMD gene. J. Sun Yet-sen University (Med. Sci.) 26, 11–15 (2005).
Flanigan, K. M., Dunn, D. M., Von Niederhausern, A., Soltanzadeh, P., Gappmaier, E., Howard, M. T. et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum. Mutat. 30, 1657–1666 (2009).
Takeshima, Y., Yagi, M., Okizuka, Y., Awano, H., Zhang, Z., Yamauchi, Y. et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J. Hum. Genet. 55, 379–388 (2010).
White, S. J., Aartsma-Rus, A., Flanigan, K. M., Weiss, R. B., Kneppers, A. L., Lalic, T. et al. Duplications in the DMD gene. Hum. Mutat. 27, 938–945 (2006).
Buzin, C. H., Feng, J., Yan, J., Scaringe, W., Liu, Q., den Dunnen, J. et al. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. Hum. Mutat. 25, 177–188 (2005).
Tuffery, G. S., Béroud, C., Leturcq, F., Yaou, R. B., Hamroun, D., Michel-Calemard, L. et al. Genotype–phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD–DMD database: a model of nationwide knowledgebase. Hum. Mutat. 30, 934–945 (2009).
Barnea, E., Zuk, D., Simantov, R., Nudel, U. & Yaffe, D. Specificity of expression of the muscle and brain dystrophin gene promoters in muscle and brain cells. Neuron 5, 881–888 (1990).
Mehler, M. F. Brain dystrophin, neurogenetics and mental retardation. Brain Res. Brain Res. Rev. 32, 277–307 (2000).
Mochizuki, H., Miyatake, S., Suzuki, M., Shigeyama, T., Yatabe, K., Ogata, K. et al. Mental retardation and lifetime events of Duchenne muscular dystrophy in Japan. Intern. Med. 47, 1207–1210 (2008).
Desguerre, I., Christov, C., Mayer, M., Zeller, R., Becane, H. M., Bastuji-Garin, S. et al. Clinical heterogengeity of Duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE 4, e4347 (2009).
Bushby, K. M., Appleton, R., Anderson, L. V., Welch, J. L., Kelly, P. & Gardner-Medwin, D. Deletion status and intellectual impairment in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 37, 260–269 (1995).
Yang, J., Li, S. Y., Li, Y. Q., Cao, J. Q., Feng, S. W., Wang, Y. Y. et al. MLPA based genotype–phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC. Med. Genet. 14, 29 (2013).
Acknowledgements
We thank the reviewers for valuable comments that largely improve the presentation of the manuscript. This work was supported by funding from the National Key Basic Research Program of China (2012CB944600), the National Key Technology R&D Program of China (2012BAI09B05), the National Natural Science Foundation of China (81270706) and the Special Fund on Scientific Research in Health Public Welfare Industry (201302001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Guo, R., Zhu, G., Zhu, H. et al. DMD mutation spectrum analysis in 613 Chinese patients with dystrophinopathy. J Hum Genet 60, 435–442 (2015). https://doi.org/10.1038/jhg.2015.43
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.43
This article is cited by
-
NGS-based targeted sequencing identified six novel variants in patients with Duchenne/Becker muscular dystrophy from southwestern China
BMC Medical Genomics (2023)
-
Mutational spectrum and phenotypic variability of Duchenne muscular dystrophy and related disorders in a Bangladeshi population
Scientific Reports (2023)
-
Clinical, muscle imaging, and genetic characteristics of dystrophinopathies with deep-intronic DMD variants
Journal of Neurology (2023)
-
Becker muscular dystrophy: case report, review of the literature, and analysis of differentially expressed hub genes
Neurological Sciences (2022)
-
Noninvasive prenatal diagnosis of duchenne muscular dystrophy in five Chinese families based on relative mutation dosage approach
BMC Medical Genomics (2021)
