
Abstract
Cataract is defined as opacity in the crystalline lens and congenital cataract occurs during the first year of life. Until now, mutations of more than 50 genes in congenital cataract have been reported with various modes of inheritance. Among them, HSF4 mutations have been reported in autosomal dominant, autosomal recessive and age-related forms of cataract. The inheritance patterns of these mutations depend on their mutational positions in HSF4: autosomal dominant or recessive mutations are respectively found either in a DNA-binding domain or in (or downstream of) hydrophobic repeats. Here we report a novel homozygous HSF4 mutation (c.521T>C, p.Leu174Pro) in two affected sibs of an Iranian consanguineous family using whole exome sequencing. The mutation is predicted as highly pathogenic by in silico analysis (SIFT, Polyphen2 and MutationTaster) and is not found in any of control databases. This mutation is located in a hydrophobic repeat of the HSF4 protein, which is consistent with the mode of inheritance as an autosomal recessive trait.
Similar content being viewed by others

Introduction
Cataract is a common ocular disorder with lens opacities or cloudiness and may be classified, based on the onset ages,1 as congenital (occurring within 1 year after birth), juvenile (occurring from 1 to 10 years of age), presenile (before 45 years) or as age-related (after 45 years) forms. In particular, congenital cataract (CC) is seen in 3–6 of 10 000 live births, and patients might have irreversible amblyopia and blindness without any early treatments.2 It is known that ~8–25% of CC cases are hereditary such as autosomal dominant, autosomal recessive and X-linked recessive modes of inheritance.2, 3 To date, mutations of at least 86 genes have been reported in cataract (Human Gene Mutation Database, Professional release 2015.1, http://www.hgmd.org), and among them, mutations of more than 50 genes cause CC.1, 4, 5
HSF4 mutations have been found in CC both with autosomal dominant and recessive modes of inheritance.6, 7, 8, 9, 10, 11, 12 Interestingly, all dominant mutations reported are located in a DNA-binding domain close to the N-terminal region of HSF4, whereas the autosomal recessive mutations reside within hydrophobic repeats (HR-A/B) or downstream of the hydrophobic repeat.6, 7, 8, 9, 10, 11, 12 Here we report a novel homozygous mutation in HSF4 with two affected patients showing autosomal recessive CC in an Iranian family. This is the second report of a missense mutation leading to autosomal recessive CC.
Materials and methods
Subjects
An Iranian family comprising of the parents, two sons and a daughter was investigated in this study. The brothers (VI-1 and VI-2 in Figure 1a) were affected with CC and nystagmus. Informed consent was obtained from the parents and blood samples were collected from the familial members at the Medical Genetics Center of Genome (Isfahan, Iran). DNA was extracted from peripheral blood leukocytes using QIAamp DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instruction. Institutional Review Boards of Yokohama City University School of Medicine approved this study.

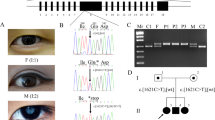
Genetic analysis of the family with congenital cataract. (a) Family pedigree with the mutation. (b) Electropherograms of affected individuals (VI-1 and VI-2) and unaffected family members (V-3, V-4 and VI-3). The HSF4 homozygous mutation (c.521T>C, p.Leu174Pro) was found in VI-1 and VI-2 (arrows). (c) Evolutionary conservation of p.Leu174 in HSF4. A full color version of this figure is available at the Journal of Human Genetics journal online.
Candidate gene identification
Whole exome sequencing was performed in one affected individual (VI-2 in Figure 1a) as described in Supplementary Methods. On the basis of the pedigree tree, we identified the autosomal recessive or X-linked recessive variants in 86 known genes, which are known to be mutated in cataract (Supplementary Table 1) and then validated the candidate variants by the Sanger sequencing method. PCR products amplified with genomic DNA as a template were sequenced on an ABI3500xL sequencer (Applied Biosystems, Foster City, CA, USA) and analyzed using Sequencher 5.0 (Gene Codes Corporation, Ann Arbor, MI, USA).
Results and discussion
Regarding whole exome sequencing, 92.5% of RefSeq coding sequences were covered by 20 or more reads in the patient, and six candidate variants in 86 known genes were identified (Table 1). Based on the hypothesis that this condition is caused by an autosomal recessive or X-linked recessive inheritance, only homozygous or compound heterozygous mutations in autosomes and hemizygous mutations in the X chromosome were analyzed. Only three homozygous variants remained: GCNT2 (NM_145655.3, c.816C>G), TRPM3 (NM_001007471.2, c.677-2->T) and HSF4 (NM_001040667.2, c.521T>C, p.Leu174Pro). GCNT2 and TRPM3 variants were unlikely to be pathogenic as they were observed in 569 and 257 of 575 individuals, respectively, in our in-house exome database. Therefore, only one homozygous missense mutation in HSF4 remained as a candidate. In contrast, this variant was not found in the following variant databases: our in-house Japanese exome database (n=575), 1000 Genomes database, Exome Aggregation Consortiumbrowser (July, 2015 accessed), or NHLBI Exome Sequencing Project (ESP6500). The Sanger sequencing revealed that the mutation co-segregated with CC affection status in the pedigree (Figure 1b). The HSF4 variant was predicted to be pathogenic based on the web-based programs (SIFT=0, Polyphen2=0.999, MutationTaster=disease causing) and the altered amino acid was highly evolutionally conserved (from Danio rerio to Human sapiens) (Figure 1c). The substitution of p.Leu174Pro is located within the hydrophobic repeats (HR-A/B) (Figure 2).13

Pathogenic HSF4 mutations in human HSF4 protein. The previously reported dominant and recessive mutations were shown above and below the HSF4 protein, respectively. The novel p.Leu174Pro was found in the hydrophobic repeats (HR-A/B) region (yellow box). The DNA-binding domain (DBD) and the downstream of hydrophobic repeat (DHR) are indicated as blue and green. A full color version of this figure is available at the Journal of Human Genetics journal online.
The HSF4 gene at 16q22.1 encodes a heat-shock transcription factor that forms the homotrimer repressing and/or activating downstream target genes such as FGF1, FGF4, FGF7 and CRYGC required in lens development.14 In HSF4 protein, three functional regions were recognized. The DNA-binding domain domain has a role for the DNA-binding mediated heat-shock element (Figure 2).6, 9, 11, 12 Missense mutations found in DNA-binding domain may have a dominant negative effect obstructing both normal and mutant HSF4 proteins.7 The HR-A/B and the downstream of hydrophobic repeat are essential for a trimeric formation and transcriptional activation of HSF4, respectively.13, 15 The novel mutation (p.Leu174Pro) identified in HR-A/B was also found in autosomal recessive CC, supporting the previous finding.7
All recessive HSF4 mutations previously reported showed significant reductions of the transcriptional activity by luciferase assay, suggesting that they were loss-of-function mutations.13, 16 Some dominant missense mutations in HSF4 (p.Ala19Asp, p.Arg73His, and p.Leu114Pro) have also shown a decreased transcriptional activity resembling recessive mutations.16 Thus, the CC-associated HSF4 mutations could be loss-of-function regardless of their inheritance pattern, including p.Leu174Pro. It was previously reported that Hsf4-null mice presented cataract with abnormal lens fiber cells, supporting that recessive (loss-of-function) HSF4 mutations lead to CC in humans.14 Therefore, the p.Leu174Pro in HSF4 is highly likely causative for CC in this family, though we did not provide any functional data.
In summary, we described here one consanguineous Iranian family showing CC caused by a novel HSF4 mutation. More mutations should be accumulated to thoroughly differentiate recessive and dominant mutations.
References
Shiels, A. & Hejtmancik, J. F. Genetics of human cataract. Clin. Genet. 84, 120–127 (2013).
Medsinge, A. & Nischal, K. K. Pediatric cataract: challenges and future directions. Clin. Ophthalmol. 9, 77–90 (2015).
Hejtmancik, J. F. Congenital cataracts and their molecular genetics. Semin. Cell Dev. Biol. 19, 134–149 (2008).
Graw, J. Congenital hereditary cataracts. Int. J. Dev. Biol. 48, 1031–1044 (2004).
Deng, H. & Yuan, L. Molecular genetics of congenital nuclear cataract. Eur. J. Med. Genet. 57, 113–122 (2014).
Bu, L., Jin, Y., Shi, Y., Chu, R., Ban, A., Eiberg, H. et al. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat. Genet. 31, 276–278 (2002).
Smaoui, N., Beltaief, O., BenHamed, S., M'Rad, R., Maazoul, F., Ouertani, A. et al. A homozygous splice mutation in the HSF4 gene is associated with an autosomal recessive congenital cataract. Invest. Ophthalmol. Vis. Sci. 45, 2716–2721 (2004).
Forshew, T., Johnson, C. A., Khaliq, S., Pasha, S., Willis, C., Abbasi, R. et al. Locus heterogeneity in autosomal recessive congenital cataracts: linkage to 9q and germline HSF4 mutations. Hum. Genet. 117, 452–459 (2005).
Ke, T., Wang, Q. K., Ji, B., Wang, X., Liu, P., Zhang, X. et al. Novel HSF4 mutation causes congenital total white cataract in a Chinese family. Am. J. Ophthalmol. 142, 298–303 (2006).
Sajjad, N., Goebel, I., Kakar, N., Cheema, A. M., Kubisch, C. & Ahmad, J. A novel HSF4 gene mutation (p.R405X) causing autosomal recessive congenital cataracts in a large consanguineous family from Pakistan. BMC Med. Genet. 9, 99 (2008).
Gillespie, R. L., O'Sullivan, J., Ashworth, J., Bhaskar, S., Williams, S., Biswas, S. et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 121, e2121–e2122 (2014).
Lv, H., Huang, C., Zhang, J., Liu, Z., Zhang, Z., Xu, H. et al. A novel HSF4 gene mutation causes autosomal-dominant cataracts in a Chinese family. G3 (Bethesda) 4, 823–828 (2014).
Merath, K., Ronchetti, A. & Sidjanin, D. J. Functional analysis of HSF4 mutations found in patients with autosomal recessive congenital cataracts. Invest. Ophthalmol. Vis. Sci. 54, 6646–6654 (2013).
Fujimoto, M., Izu, H., Seki, K., Fukuda, K., Nishida, T., Yamada, S. et al. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 23, 4297–4306 (2004).
Akerfelt, M., Morimoto, R. I. & Sistonen, L. Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555 (2010).
Enoki, Y., Mukoda, Y., Furutani, C. & Sakurai, H. DNA-binding and transcriptional activities of human HSF4 containing mutations that associate with congenital and age-related cataracts. Biochim. Biophys. Acta 1802, 749–753 (2010).
Acknowledgements
The authors thank the members of the family for their participation in this study. This work was supported in part by a grant for Research on Measures for Intractable Diseases, a grant for Comprehensive Research on Disability Health and Welfare, the Strategic Research Program for Brain Science (SRPBS) from Japan Agency for Medical Research and Development (AMED); a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT); Grants-in-Aid for Scientific Research (A and B), a Grant-in-Aid for Research Activity start-up from the Japan Society for the Promotion of Science (JSPS); the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency (JST); the Takeda Science Foundation; the Yokohama Foundation for Advancement of Medical Science; and the Hayashi Memorial Foundation for Female Natural Scientists.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Behnam, M., Imagawa, E., Chaleshtori, A. et al. A novel homozygous mutation in HSF4 causing autosomal recessive congenital cataract. J Hum Genet 61, 177–179 (2016). https://doi.org/10.1038/jhg.2015.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.127
This article is cited by
-
Loss of FYCO1 leads to cataract formation
Scientific Reports (2021)
-
Genetic landscape of isolated pediatric cataracts: extreme heterogeneity and variable inheritance patterns within genes
Human Genetics (2019)
-
A novel missense mutation in HSF4 causes autosomal-dominant congenital lamellar cataract in a British family
Eye (2018)
-
A zebrafish model of foxe3 deficiency demonstrates lens and eye defects with dysregulation of key genes involved in cataract formation in humans
Human Genetics (2018)
