
Abstract
The locus control region (LCR) is a genetic region that regulates the expression of the β-globin locus (HBB locus). This region is composed of several DNase I hypersensitive sites (HSs) in which the regulatory functions of the LCR may reside. To date, some individuals bearing deletions of several HSs or even the complete LCR have been described. Although the globin genes of the HBB locus are intact, most of these patients suffer thalassemia due to the reduced expression of such genes. The LCR and the HSs forming it have been thoroughly studied in different genetic models. However, seemingly contradictory results are often obtained. Here, we describe the first deletion found in humans exclusively affecting the HS3 element of the LCR. The adult carrying this deletion shows very mild hematological modifications, indicating that HS3 deletion does not severely impair the β-gene expression. Our results also reveal limitations of the murine models when studying the native mouse genes for understanding human diseases like thalassemias.
Similar content being viewed by others


Main
The β-globin locus (HBB locus) contains several globin genes that are expressed sequentially during human ontogeny to form part of hemoglobin. The main regulatory element of this locus is known as the locus control region (LCR). It is well proven that the LCR mediates a series of epigenetic and structural changes in the HBB locus that allow high expression of its genes. The LCR has such an important role that humans carrying a deletion of this element are clinically identical to individuals carrying deletions of the entire HBB locus. Globin genes on that chromosome are not expressed, and a phenotype known as ɛγδβ thalassemia results.1 The LCR is characterized by five DNase I hypersensitive sites (HS1–HS5). Each HS can be bound by different proteins that are involved in the rearrangement of the chromatin or in transcriptional activation, and it is thought that the LCR functions reside in these genetic elements.2, 3
There is still much controversy in explaining the role each HS plays in the activation of globin genes. This is mainly due to the disparity in the results found when analyzing the effect of each HS in different biological models. Studies on transgenic mice with a human HBB locus have shown that the deletion of HS3 affects the expression of the ɛ and γ genes during embryonic and fetal development, whereas the expression of β in the adult stage is not affected.2, 4 Interestingly, when examining the murine HBB locus with HS3 deletions, the opposite effect has been observed; the adult β globin genes (βmaj and βmin) are the only genes with considerably reduced expression.5
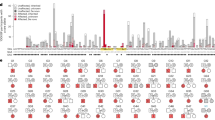
The 34-year-old man carrier of the HS3 deletion presented Hb levels of 15.2 g dl−1, MCV 79.8 fl, MCH 26.9 pg and serum ferritin 197 ng ml−1. Familiar studies were not available; therefore it was not possible to assess if the HS3 deletion was inherited. Hemoglobin analysis by high-performance liquid chromatography revealed HbF 0.2% and HbA2 2.1%. The deletion was found by multiplex ligation-dependent probe amplification (kit P102-B2 HBB, MRC-Holland, Amsterdam, The Netherlands) (Figure 1a). To complete the genetic study, multiplex ligation-dependent probe amplification for the HBA locus and direct DNA sequencing of the α and β globin genes were performed as previously described.6 All these tests produced negative results.

Genetic findings in this study. (a) MLPA profile of the HBB loci of the individual studied here. This profile indicates the existence of a heterozygous deletion (ratio ∼0.5) in the HS3 region. (b) Alignment of the nucleotide sequences containing the deletion (HS3 del), the normal sequence located upstream of the 5′ breakpoint (5′N) and the normal sequence located downstream of the 3′ breakpoint (3′N). Gap PCR followed by sequencing was performed to define the deletion’s breakpoints of the HS3 deletion. The 5′ end occurs after nucleotide 11834 or 11835 within the HBB locus (NCBI reference sequence: NG_000007.3). The 3′ end occurs inside an Alu sequence, after nucleotide 13826 or 13827. The alignment does not allow us to determine if the nucleotide G inside the box belongs to the normal sequence in the 5′ position (11835) or in the 3′ position (13827). In any case, the deletion removes 1992 nucleotides. The primers used for the amplification of the DNA containing the deletion were 5′-GGGGTGGTGGTTTTGATTGC-3′ (forward) and 5′-TGG GATGGGGGAAAAGAATGT-3′ (reverse). The forward primer was used for subsequent sequencing of the amplified fragments.
Analysis of the HS3 deletion breakpoints showed that the deletion was 1992 bp in size (Figure 1b). This variant was submitted to HbVar database7 and has been denoted as NG_000007.3:g.(11834_11835)_(13826_13827)del1992.
To date, a series of human deletions affecting the LCR has been described. Deletions of both HS2 and HS3 were sufficient to trigger an ɛγδβ thalassemia-like phenotype, and therefore it appeared that the β gene required these elements of the LCR to be expressed at high levels. Such observations were supported by some studies on the control of HBB locus expression, which described a major role for HS3 and a more minor role for HS2 in promoting the expression of the β gene.3, 8 However, no deletion in humans affecting only one of these elements that could clarify the role of these HSs has been found to date. The genetic alteration presented here is relevant because it is the first deletion of HS3 observed in the HBB human locus (Figure 2).

Comparison of deletions described in the human LCR and their associated phenotypes. The deletions affecting the entire LCR produce ɛγδβ thalassemia. The HS1 deletion is not associated with hematological changes,16 which suggests that this element is not involved in the regulation of adult globin gene expression. The Hispanic deletion (HS2–HS5) was found in a patient with ɛγδβ thalassemia who was also a carrier of a mutation in HbS. However, no HbS trace was detected in the analysis.1 This result indicates that the HbS mutation was in cis with the Hispanic deletion, that the latter stops the expression of gene βS located in the same locus, and that the required sequences for high expression of gene β are found among the deleted HSs (HS2–HS5). The Tennessean deletion (HS1–HS3) also causes ɛγδβ thalassemia, which demonstrates several concepts. First, retaining HS4 and HS5 does not improve the phenotype. Second, HS2 and HS3 may be mainly responsible for enhancing the expression of gene β.9 However, our findings clearly show that HS3 deletion does not have the expected effect on the expression of gene β because its carrier does not exhibit the symptoms of ɛγδβ thalassemia.
Surprisingly, the HS3 deletion is associated with a nearly normal phenotype in the carrier, who shows no anemia and only suffers very mild microcytosis (Hb=15.2 g dl−1; MCV=79.8 fl; MCH=26.9 pg). These values are very different from those found in adult carriers of LCR deletions (patients with ɛγδβ thalassemia), which typically exhibit MCVs and MCHs around 60 fl and 20 pg, respectively.9 Given the exhaustive genetic study performed here, any condition that could mask a low production of β globin (for example, co-inherited α-thalassemia) was excluded in the carrier. Together, these results clearly indicate that the sequences removed by the HS3 deletion are not necessary for the correct and high expression of the β gene. We cannot rule out the possibility of a small reduction in the expression of δ and β in the subject carrying the deletion of HS3, as the subject’s HbA2 (2.1%) and MCH (26.9 pg) are close to the lower limits of normality in our laboratory (HbA2 normal range: 2.2–3.4%; MCH normal range: 27–34 pg). It is known that, like the HS3 deletion, silent β-thalassemia mutations cannot severely affect the β-globin gene expression and display nearly normal hematological indices. However, the coinheritance of a silent β-globin mutation and a severe β-globin mutation results in a clinical phenotype of mild non-transfusion-dependent β-thalassemia intermedia.10 Therefore, it is possible that coinheritance of the HS3 deletion with a severe β thalassemia mutation in TRANS could lead to a β-thalassemia intermedia phenotype.
Our findings support previous observations in transgenic mice containing the human HBB locus. The researchers found that several types of deletions in HS3 had no substantial effect on the expression of the β gene in adults.11 These results have not been reproduced in any of the studies that have been performed on the native HBB locus of the mouse. Unlike the results for the human locus, the loss of any HS in the native locus of the mouse causes a decrease of ∼30% in the expression of adult globin genes.5, 12, 13, 14 Such a discrepancy between human and murine regulatory elements has also been documented previously in the HBA locus. The deletion of the element HS-26 (HS-40 in humans) has a minimal effect on the phenotype of the mouse, but results in a clear thalassemic phenotype in humans.15 All of the above results demonstrate the limited value of studies on the regulation of native mouse genes to understanding the molecular mechanisms causing human diseases such as thalassemia.
Other interesting observations obtained in transgenic mice showed that HS3 may behave as a specific enhancer of embryonic and fetal genes.2, 4 We know that the subject studied here did not show severe anemia in the perinatal stage. Unfortunately, we do not have precise hematological data at birth when fetal globins are still highly synthesized, which could have helped us to assess whether HS3 has specificity for the activation of fetal globin genes in humans.
Finally, HS deletions found in humans so far indicate that HS2 is the main regulator of β gene expression or that HS2 and HS3 have redundant functions when controlling the expression of this gene. Finding a deletion restricted to HS2 in humans could resolve this issue. In summary, we have shown that human HS3 deletion has an unexpected mild effect on red cell phenotype in adults, although we cannot rule out an important role for this element in the control of globin gene expression in other stages of human development.
References
Driscoll, M. C., Dobkin, C. S. & Alter, B. P. Gamma delta beta-thalassemia due to a de novo mutation deleting the 5′ beta-globin gene activation-region hypersensitive sites. Proc. Natl Acad. Sci. USA 86, 7470–7474 (1989).
Fraser, P., Pruzina, S., Antoniou, M. & Grosveld, F. Each hypersensitive site of the human beta-globin locus control region confers a different developmental pattern of expression on the globin genes. Genes Dev. 17, 106–113 (1993).
Kim, S., Kim, Y. W., Shim, S. H., Kim, C. G. & Kim, A. Chromatin structure of the LCR in the human β-globin locus transcribing the adult δ- and β-globin genes. Int. J. Biochem. Cell. Biol. 44, 505–513 (2012).
Peterson, K. R., Clegg, C. H., Navas, P. A., Norton, E. J., Kimbrough, T. G. & Stamatoyannopoulos, G. Effect of deletion of 5′HS3 or 5'HS2 of the human beta-globin locus control region on the developmental regulation of globin gene expression in beta-globin locus yeast artificial chromosome transgenic mice. Proc. Natl Acad. Sci. USA 93, 6605–6609 (1996).
Hug, B. A., Wesselschmidt, R. L., Fiering, S., Bender, M. A., Epner, E., Groudine, M. et al. Analysis of mice containing a targeted deletion of beta-globin locus control region 5′ hypersensitive site 3. Mol. Cell Biol. 16, 2906–2912 (1996).
De la Fuente-Gonzalo, F., Baiget, M., Badell, I., Ricard, P., Vinuesa, L., Martínez-Nieto, J. et al. Study of three families with Hb Agrinio [α29(B10)Leu→Pro, CTG>CCG (α2)] in the Spanish population: three homozygous cases. Hemoglobin 36, 526–532 (2012).
Patrinos, G. P., Giardine, B., Riemer, C., Miller, W., Chui, D. H. K., Anagnou, N. P. et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Res. 32, D537–D541 (2004).
Ellis, J., Tan-Un, K. C., Harper, A., Michalovich, D., Yannoutsos, N., Philipsen, S. et al. A dominant chromatin-opening activity in 5′ hypersensitive site 3 of the human beta-globin locus control region. EMBO J. 15, 562–568 (1996).
Koenig, S. C., Becirevic, E., Hellberg, M. S. C., Li, M. Y., So, J. C. C., Hankins, J. S. et al. Sickle cell disease caused by heterozygosity for Hb S and novel LCR deletion: report of two patients. Am. J. Hematol. 84, 603–606 (2009).
Maragoudaki, E., Kanavakis, E., Traeger-Synodinos, J., Vrettou, C., Tzetis, M., Metaxotou-Mavrommati, A. et al. Molecular, haematological and clinical studies of the -101 C —> T substitution of the beta-globin gene promoter in 25 beta-thalassaemia intermedia patients and 45 heterozygotes. Br. J. Haematol 107, 699–706 (1999).
Peterson, K. R., Fedosyuk, H. & Harju-Baker, S. LCR 5′ hypersensitive site specificity for globin gene activation within the active chromatin hub. Nucleic Acids Res. 40, 11256–11269 (2012).
Bender, M. A., Roach, J. N., Halow, J., Close, J., Alami, R., Bouhassira, E. E. et al. Targeted deletion of 5′HS1 and 5′HS4 of the beta-globin locus control region reveals additive activity of the DNaseI hypersensitive sites. Blood 98, 2022–2027 (2001).
Hu, X., Bulger, M., Bender, M. a., Fields, J., Groudine, M. & Fiering, S. Deletion of the core region of 5′ HS2 of the mouse beta-globin locus control region reveals a distinct effect in comparison with human beta-globin transgenes. Blood 107, 821–826 (2006).
Bender, M. a., Ragoczy, T., Lee, J., Byron, R., Telling, A., Dean, A. et al. The hypersensitive sites of the murine β-globin locus control region act independently to affect nuclear localization and transcriptional elongation. Blood 119, 3820–3827 (2012).
Anguita, E., Sharpe, J. A., Sloane-Stanley, J. A., Tufarelli, C., Higgs, D. R. & Wood, W. G. Deletion of the mouse alpha-globin regulatory element (HS -26) has an unexpectedly mild phenotype. Blood 100, 3450–3456 (2002).
Kulozik, A. E., Bail, S., Bellan-Koch, A., Bartram, C. R., Kohne, E. & Kleihauer, E. The proximal element of the beta globin locus control region is not functionally required in vivo. J. Clin. Invest. 87, 2142–2146 (1991).
Acknowledgements
This work was partially supported by Fondo de Investigación Sanitaria (FIS, PI12/01068 to P Ropero) and co-financed by the Asociación Madrileña de Hematología y Hemoterapia (AMHH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nieto, J., Villegas, A., De La Fuente-Gonzalo, F. et al. Heterozygosity for deletion of hypersensitive site 3 in the human locus control region has an unexpected minor effect on red cell phenotype. J Hum Genet 59, 585–587 (2014). https://doi.org/10.1038/jhg.2014.76
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.76
