
Abstract
Oculofaciocardiodental (OFCD) syndrome is a rare X-linked dominant condition. Mutations in BCOR have been described as causal in OFCD syndrome. Almost all BCOR mutations result in premature termination codons (PTCs); therefore, nonsense-mediated mRNA decay (NMD) might have an important role in pathogenesis. The purpose of this study was to identify BCOR mutations in two OFCD patients, if it present, and to clarify the pathogenesis of radiculomegaly using one OFCD patient’s pulp and periodontal ligament (PDL) cells. In our genetic analysis, two novel BCOR mutations were found. We also examined the transcript levels and the effects of NMD using cultured pulp and PDL cells from one affected patient. BCOR expression was normal in pulp but reduced in PDL cells, which is consistent with the higher rates of NMD in PDL cells. The mutant PDL cells had unstable mutant transcripts and proliferated faster than did wild-type cells, but mutant pulp cells appeared normal by these measures. In summary, the nonsense and frameshift mutations, which introduce PTCs, were found to contribute to OFCD syndrome in our two patients. Furthermore, in PDL cells, the mutation resulting in a PTC corresponded to greater NMD, unstable mutant transcripts and increased cell proliferation, which may contribute to hyperactive root formation.
Similar content being viewed by others

Introduction
Oculofaciocardiodental (OFCD, MIM 300166) syndrome is a rare, X-linked dominant hereditary trait with skewed X inactivation1 in heterozygous females. Males with this syndrome cannot survive because of embryonic lethality.2, 3, 4 OFCD syndrome is characterized by microphthalmia, congenital cataracts, facial dysmorphic features, congenital heart defects and dental anomalies.1, 3, 4, 5, 6, 7 Most affected OFCD patients have remarkable dental anomalies, including radiculomegaly with prolonged dental roots and widely open apices, most often in the canine roots.4, 6, 7, 8 Recently, mutations in the BCOR gene, which encodes the BCL6 corepressor, have been found to cause OFCD syndrome.3, 5 The BCOR gene is located on the short (p) arm of the X chromosome at the position 11.4. BCOR is a transcriptional corepressor that was originally identified by its ability to interact with the site-specific transcriptional repressor BCL-6.9
Previous studies have reported that, owing to the phenotypic overlap, OFCD and Lenz microphthalmia syndrome are allelic disorders.3, 5, 7 Lenz microphthalmia syndrome is an X-linked recessive condition comprising microphthalmia/anophthalmia with mental retardation, malformed ears, skeletal, renal and urogenital anomalies. Two genetic loci that can cause what is currently considered Lenz microphthalmia syndrome have been reported: one is located at Xq27-q28 (MCOPS1; MIM 309800),10 and the second is located at Xp11.4 (MCOPS2; MIM 300166).3, 11 The MCOPS2 form of Lenz microphthalmia syndrome has been shown to be caused by mutations in the BCOR gene. A single missense change, c.254C>T (p.P85L), in BCOR has been found in patients of a family with MCOPS2.3 However, BCOR mutations are not a major cause of X-linked microphthalmia in males.5
A previous study in mouse embryos showed that Bcor expression was observed in the eye, brain, neural tube and branchial arches and that the expression levels correlated with the tissues affected in OFCD patients.12 BCOR has multiple roles in the complex process of human development, including the correction of lateral organization.13 Moreover, BCOR maintains tissue homeostasis and gene silencing through epigenetic mechanisms.2, 12, 14 BCOR expression has been detected in both the dental epithelium and mesenchyme during tooth development in early embryogenesis, and BCOR expressed in the mesenchyme has an important role in proper tooth formation.15 Moreover, mutant BCOR increased the osteo-dentinogenic potential of mesenchymal stem cells (MSCs) by inducing AP-2α, and it increased the cell proliferation rate.2 However, the mechanism of hyperactive root formation in OFCD syndrome is still unclear.
Almost all of the mutations identified in individuals with OFCD syndrome result in predicted premature termination codons (PTCs) and have similar phenotypic consequences.3, 4, 5, 7 A mutation that leads to PTC will be transcribed into mRNA and then translated into a truncated protein. The mRNA will be degraded by a protective mechanism in the cell called nonsense-mediated mRNA decay (NMD) to shield cells from dominant-negative or deleterious gain-of-function activities of mutant proteins. The efficiency of NMD is an inherent characteristic and varies among different cells.16, 17 NMD may exert a beneficial, neutral or harmful effect, depending on the location of the PTC in the transcript and the properties of the truncated protein. NMD can also contribute to a disease phenotype when it inhibits the expression of partially functional proteins.18 According to previous studies of BCOR mutations, it is likely that NMD prevents the truncated proteins from being expressed in vivo.3, 6 However, NMD was not confirmed because of the severely skewed X-inactivation in blood leukocytes.3, 13 Here, for the first time, we report alterations in the transcript levels of BCOR due to NMD in cultured pulp and periodontal ligament (PDL) cells from one affected patient.
In our study, we identified novel mutations in the BCOR genes of two affected OFCD patients. In addition, PDL and pulp tissue were harvested from a first premolar of one affected patient that was extracted for orthodontic purposes and then cultured. We performed an analysis of NMD, RNA stability and cell proliferation on these two cell types, which confirmed our hypothesis that the mutant RNAs were degraded by NMD mechanisms leading to haploinsufficiency of the BCOR protein, and compromised BCOR-mediated regulatory activities in root formation.
Materials and methods
Subjects
Two female OFCD patients (aged 27 years and 18 years) and three healthy subjects were patients of the Department of Orthodontics, Dental Hospital, Tokyo Medical and Dental University, Japan. The healthy patient 1’s parents and the healthy patient 2’s mother were also included in the genetic analysis. The clinical findings and orthodontic analysis of both OFCD patients at the first visit were previously described by Tsukawaki et al.19 The diagnosis of OFCD syndrome was based on a distinct pattern of eye, craniofacial, heart and dental anomalies. Informed consent was obtained from both patients and their parents. The human subject research described here was approved by the Ethical Review Committee of the Tokyo Medical and Dental University.
Mutation analysis
Genomic DNA was extracted from buccal swabs with the QIAamp DNA Mini kits (Qiagen, Germantown, MD, USA) according to the manufacturer’s protocol. Coding exons and flanking introns of the BCOR gene (15 exons: GenBank accession no. AY316592) were amplified by conventional PCR using KOD-Plus (TOYOBO, Lifescience, Tokyo, Japan). The primer sequences and PCR conditions used for the BCOR gene were previously described.7 All novel mutations that we found were submitted to Leiden Open Variation Database (http://databases.lovd.nl/shared/genes/BCOR). The presence of a PCR product was verified by gel electrophoresis of the PCR product on a 1.5% agarose gel stained with ethidium bromide. Thereafter, the PCR products were directly sequenced with the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Life Technologies, Foster City, CA, USA) on a Perkin Elmer PE 9700 Thermal Cycler. The mutations were evaluated for their disease-causing potential based on sequence alterations by the ‘Mutation Taster’ (http://www.mutationtaster.org/)20 and confirmed with the Japanese Single Nucleotide Polymorphisms Database (http://snp.ims.u-tokyo.ac.jp/index.html).
Cell culture
The first premolars of patient 2 and three healthy subjects were extracted for orthodontic reasons. The PDL surrounding the apical 1/3 of the root and apical pulp tissues were harvested using an aseptic technique, as previously described.21, 22, 23 The cells were cultured in αMEM (Wako, Osaka, Japan) supplemented with 10% fetal bovine serum (Gibco, Life Technologies, Paisley, UK) and penicillin-streptomycin (Gibco). All cultures were maintained in a humidified, 5% CO2 atmosphere at 37 °C. No cells were passaged more than five times (P5). Genomic DNA was extracted from both pulp and PDL cells to confirm the sequence of the BCOR gene.
RNA preparation
Total RNA was isolated from cultured pulp and PDL cells from the patient and three normal controls using ISOGEN (Nippon Gene, Toyama, Japan). Reverse transcription was performed using 1 μg of total RNA and the QuantiTect Reverse Transcription Kit (Qiagen) in a total volume of 20 μl according to the manufacturer’s instructions.
Characterization of the cultured cells
Transcript levels of vimentin (VIM) and keratin 14 (KRT14) were measured and used as markers of mesenchymal and epithelial cells, respectively. Total RNA extraction, cDNA synthesis and conventional PCR were performed. The primers used were as follows: VIM, forward, 5′-AGGTGGACCAGCTAACCAAC-3′ and reverse, 5′-AGCATCTCCTCCTGCAATTT-3′; KRT14, forward, 5′-ATTGAGAGCCTGAAGGAGGA-3′ and reverse, 5′-ATTGACATCTCCACCCACCT-3′; and β-ACTIN, forward, 5′-CATGTACGTTGCTATCCAGGC-3′ and reverse, 5′-CTCCTTAATGTCACGCACGAT-3′. The PCR cycling conditions for VIM, KRT14 and β-ACTIN consisted of an initial denaturation step at 94 °C for 3 min, 30 cycles of amplification (15 s at 94 °C; 10 s at 55 °C; 45 s at 72 °C) and a final extension step at 72 °C for 4 min. Gel electrophoresis was performed for the semi-quantitative evaluation of gene expression.
Real-time PCR
Each cDNA was used as a template for the quantitative analysis of BCOR mRNA expression with a 7300 Real-Time PCR system (Applied Biosystems). The real-time PCR reactions were performed using the Power SYBR Green PCR master mix (Applied Biosystems), according to the manufacturer’s instructions. The total RNA input into each reaction was constant. The primers used were as follows: BCOR, forward, 5′-TCCTCGACTCGCCAAATAAG-3′ and reverse, 5′-TGGCATAGTGCTTGTGGAAC-3′; β-ACTIN, forward, 5′-CATGTACGTTGCTATCCAGGC-3′ and reverse, 5′-CTCCTTAATGTCACGCACGAT-3′. The real-time PCR conditions are available on request.
Analysis of NMD
We examined the levels of mutant transcripts following treatment with cycloheximide (CHX), a general inhibitor of translation elongation. Because NMD is translation-dependent, CHX treatment leads indirectly to the inhibition of NMD.16, 24 Cells were grown in two 10-cm-diameter cell culture dishes until they reached 70% confluence. The medium was changed and replaced with 5 mg CHX (Wako) dissolved in 10 ml complete medium (for a final CHX concentration of 500 μg ml−1) and then incubated at 37 °C. Total RNA was extracted from the cells at 0 and 6 h after adding CHX, and then BCOR expression was evaluated using real-time PCR. The experiments were performed in triplicate.
Analysis of mRNA stability
Cells grown in three 10-cm-diameter cell culture dishes were treated with 1 μg ml−1 of actinomycin D (Wako) and incubated at 37 °C. Total RNA was isolated from the cells at 0, 3 and 6 h after adding actinomycin D, and BCOR expression was evaluated using real-time PCR. The experiments were performed in triplicate.
Cell proliferation rate
To monitor cell proliferation, MTT assays were performed. Pulp and PDL cells from the patient and normal controls were plated in a 96-well plate with a volume of 100 μl per well (7500 total cells). Six wells were allotted per cell type, and wells with complete medium served as a negative control. All cells were incubated at 37 °C. Pulp cells were incubated for 1, 2, 3 or 4 days, and PDL cells were incubated for 24, 36 or 48 h. Subsequently, each well was incubated with 20 μl of 5 mg ml−1 MTT (Sigma, St Louis, MO, USA) at 37 °C for 3.5 h. The MTT solution was removed, and 150 μl MTT solvent (4 mM HCl, 0.1% Nonidet P-40 all in isopropanol) was added. The plate was covered with aluminum foil, and the cells were agitated on an orbital shaker for 15 min, after which time the absorbance was read at 590 nm with a reference filter of 620 nm using a Bio-Rad model 680 microplate reader (Bio-Rad, Hercules, CA, USA).
Statistical analysis
The SPSS version 21 software package was used for all statistical analyses. All experimental data were reported as the mean±SD. Mean values of continuous variables were compared using a two-tailed t-test for independent samples. P-values of less than 0.05 and 0.01 were considered significant and highly significant, respectively.
Results
Clinical findings
The current panoramic radiographs of the two affected individuals and the pedigree of each patient are shown in Figures 1a, b, d and e. Patient 1 is the first child of her family, and her parents and younger sister is normal. Patient 2 is the second child of her family, and her parents and elder brother is normal. Therefore, both cases are sporadic mutations. In this study, more details of the dental phenotypes of the patients were examined. The record of an oral examination of patient 1 (first visit: 16Y6M) showed nine missing and six impacted teeth, persistent primary teeth, delayed secondary dentition, radiculomegaly, malformed teeth and cleft palate. Patient 2 (first visit: 10Y9M) had similar but less severe oral phenotypes. She had only two missing teeth, one impacted tooth and submucous cleft palate. The details of the dental phenotypes at the initial stage are summarized in the diagrams in Figures 1c and f.

Family pedigree, current panoramic radiograph and summarized dental phenotypes of the two patients. The results of patient 1 are shown in a–c, and the results of patient 2 are shown in d–f. (a) Family pedigree of patient 1. The arrow indicates patient 1. (b) Current panoramic radiograph of patient 1 showing remarkably long roots in many teeth, especially in the anterior and premolar region. (c) Summary of the dental phenotype at the initial stage of patient 1. (d) Family pedigree of patient 2. The arrow indicates patient 2. (e) The current panoramic radiograph of patient 2 also shows remarkably long roots and open apices in many teeth in the anterior and premolar region. (f) Summary of the dental phenotype at the initial stage of patient 2.
Mutation analysis
Genetic analysis showed that patient 1 harbored a heterozygous intronic polymorphism, c.166-55G>A, and a heterozygous nonsense mutation, c.*4794G>A (p.W1598*). The results of the healthy parents of patient 1 showed that the father and mother harbored the same intronic polymorphism, homozygous in the father and heterozygous in the mother, but not the nonsense mutation (Figures 2a and b). This intronic polymorphism was reported in the Japanese Single Nucleotide Polymorphisms Database, reference as rs6610384. Patient 2 harbored a heterozygous frameshift mutation, c.3668delC (p.S1223Wfs*15), whereas her healthy mother did not have any mutations (Figure 2c). Disease-causing potential analyzed by Mutation Taster showed that the nonsense and frameshift mutations are predicted to cause disease, and no single nucleotide polymorphisms in the altered region were found. These two mutations had not been previously reported. The positions of the mutations and polymorphism relative to the BCOR exons and protein are depicted in Figure 2d.

Genetic analysis. (a and b) Patient 1 had an intronic polymorphism, c.166-55G>A and a nonsense mutation, c.4794G>A, which caused the truncated protein p.W1598*. However, her parents harbored only the intronic polymorphism. (c) Patient 2 had a frameshift mutation, c.3668delC, which caused the truncated protein p.S1223Wfs*15, whereas her mother’s sequence was normal. (d) The positions of the variations in BCOR relative to the exons and the protein. The variations in patient 1 are shown as solid-line arrows and boxes. The variation in patient 2 is shown as a dotted-line arrow and box. Bcl6, the region that interacts with Bcl6; AF9, the region that interacts with AF9, the common mixed lineage leukemia gene fusion partner; NSPC1, the region that interacts with nervous system polycomb1; Ankyrin, ankyrin repeats that are involved in protein–protein interactions and protein structure.
Cell culture and characterization of cultured cells
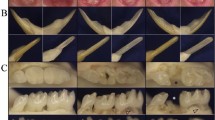
The first premolar from patient 2 was 39.2 mm in length, approximately two times longer than normal (Figure 3a). As a comparison, the normal tooth length of the lower first premolar is reported to be 22.5 mm.25 Cultured mutant pulp and PDL cells harvested from patient 2’s tooth are pictured in Figures 3b and c. Cellular BCOR gene sequences were identical to those from the buccal swab (data not shown). VIM was strongly expressed in mutant pulp, but KRT14 was not, suggesting that most of the cultured mutant pulp cells were mesenchymal and that few were epithelial cells. By contrast, VIM and KRT14 were both highly expressed in the mutant PDL cells and more highly than in mutant pulp cells. We conclude that cultured mutant PDL cells include both mesenchymal and epithelial cells and could be expected to have more mesenchymal and epithelial characteristics than cultured mutant pulp. The PCR results are shown in Figure 3d.

Cultured cells and NMD analysis. (a) Lower first premolar of patient 2. It was 39.2 mm in length. (b and c) Cultured pulp and PDL cells harvested from patient 2’s tooth. (d) VIM and KRT14 expression in pulp and PDL cells. (e) BCOR expression in pulp cells and PDL cells. (f) BCOR differential expression in pulp cells. (g) BCOR differential expression in PDL cells. (h) BCOR cDNA sequences of cultured cells before and after adding CHX. WT-Pulp, wild-type pulp; MT-Pulp, mutant pulp; WT-PDL, wild-type PDL; MT-PDL, mutant PDL; CHX(−), before adding CHX; CHX(+), after adding CHX. *P<0.05, **P<0.01.
BCOR expression
BCOR expression was detected in pulp and PDL in both wild-type and mutant cultures. BCOR expression was significantly higher in PDL cultures than in pulp cultures for both wild-type and mutant cultures. In PDL cultures, BCOR expression was significantly lower in mutant than wild-type cultures. The expression did not differ between mutant and wild-type pulp cultures. The results are shown in Figure 3e.
Analysis of NMD
Wild-type and mutant cells derived from both pulp and PDL showed increased BCOR transcript levels after the inhibition of translation elongation by the addition of CHX. The differential expression of BCOR was determined by dividing post-CHX transcript levels by pre-CHX transcript levels. This value reflected the fold change in transcript levels due to the inhibitory effect of CHX (fold induction). The differential expression of BCOR did not differ between mutant and normal pulp but was significantly higher in mutant PDL than wild-type PDL, as shown in Figures 3f and g. However, in both cell types, NMD had a partial effect on transcript levels as indicated by the detection of all heterozygous sequences in the cDNA both before and after adding CHX (Figure 3h).
Analysis of RNA stability
Mutant BCOR transcripts in the PDL cells of patient 2 degraded within 3 h after the addition of actinomycin D and were more unstable than were those of the wild type. In pulp cells, by contrast, the mutant BCOR transcript level was not significantly different from normal. The results are shown in Figures 4a and b.

BCOR transcript stability and cell proliferation rate. (a) BCOR transcript stability in pulp cells. (b) BCOR transcript stability in PDL cells. (c) Proliferation rate of pulp cells. (d) Proliferation rate of PDL cells. WT, wild-type; MT, mutant. *P<0.05, **P<0.01.
Cell proliferation rate
The MTT assay showed that the mutant PDL cells proliferated faster than the wild-type cells but that the mutant pulp cell proliferation rate was likely normal, as shown in Figures 4c and d.
Discussion
The two patients were diagnosed with OFCD syndrome based on a distinct pattern of eye, craniofacial, heart and dental anomalies. According to their family pedigrees, their parents and siblings are healthy, so both patients had sporadic mutations. Patient 1 had a heterozygous intronic polymorphism, c.166-55G>A, and a heterozygous nonsense mutation, c.*4794G>A (p.W1598*). The intronic polymorphism was also found in her healthy parents: the homozygous father and heterozygous mother, and among Japanese people as determined by the Japanese Single Nucleotide Polymorphisms Database, reference as rs6610384. The population diversity of this single nucleotide polymorphism showed that Japanese population in Tokyo have allele C=33.1% and allele T=66.9% in the reverse stand sequence. Therefore, this single nucleotide polymorphism is non-pathogenic and can be found in normal population. Patient 2 was found to have a heterozygous frameshift mutation, c.3668delC (p.S1223Wfs*15), but her healthy mother did not. Furthermore, disease-causing potential analyzed by Mutation Taster showed that the nonsense and frameshift mutations are predicted to cause disease, and no single nucleotide polymorphisms in the altered region were found. OFCD syndrome is presumed to have lethal effects on affected male. Previous studies showed that all affected individuals are females, with several incidences of mother–daughter transmission.1, 5, 26 There is no report of males developing this condition.4, 8 However, the patient 2’s father was not included in our experiment, we assumed that he does not have any pathogenic mutations in BCOR gene regarding his normal phenotypes. In addition, a single missense change, c.254C>T (p.P85L) in BCOR that has been found in patients with MCOPS2 were not found in our patients. Taken together, these data allow us to conclude that the nonsense mutation in patient 1 and the frameshift mutation in patient 2 contributed to OFCD syndrome; both mutations are pathogenic. A variation in phenotype severity was observed, suggesting that further studies are needed to clarify the genotype–phenotype relationship.
To date, a total of 34 mutations in BCOR have been found, most of which result in premature termination of the protein with deletion of the carboxy-terminal domain;3, 6, 7 therefore, NMD might have an important role in pathogenesis. Interestingly, the efficiency and sensitivity of the NMD mechanism differ by cell type.16, 17 The effect of NMD on a disease phenotype depends on both the affected gene and the location of the disease-causing PTC.18 A minimal distance of 50–55 nt between the PTC and the downstream intron is important for efficient nonsense decay.18, 27, 28, 29 If the mutation was located at fewer than 50–55 nt before the last intron, NMD would not occur, and the truncated protein would be translated. NMD may exert a beneficial, neutral or harmful effect, depending on the location of the PTCs in the transcript and the properties of the truncated protein. If the truncated mutant protein has dominant-negative activity, NMD can confer a protective effect that benefits heterozygous carriers of PTCs. By contrast, NMD can also contribute to a disease phenotype when it inhibits the expression of partially functional proteins.18 In the case of BCOR mutations, all of the affected patients have similar phenotypes, irrespective of the location of the mutation, although the phenotypes differ in severity. This observation suggests that the pathogenesis might be haploinsufficiency rather than a dominant negative effect of mutant proteins. It was reported that the truncated OFCD protein in affected patients showed repressor effects equivalent to those of wild-type BCOR.3 Therefore, we hypothesized that the mutant BCOR proteins would have partially functional effects and that the abnormal phenotypes observed in the affected tissues were the result of NMD of mutant RNAs, which would result in haploinsufficiency.
Different tissues have different NMD efficiencies. This situation could result in a selective effect in which only NMD-sensitive tissues become abnormal, which is consistent with the typical characteristics of OFCD syndrome. We focused our study on the tooth root, which becomes hyperactive and much longer than normal in OFCD-affected patients. During tooth root development, all functional hard tissues are formed by three types of cells: Hertwig’s epithelial root sheath, dental papilla mesenchymal and dental follicle cells, which form developing apical complexes.30, 31, 32 Developing apical complexes are located in the apical region of the developing tooth and can develop into an entire tooth root in vitro, without a crown.33 Mice studies showed that Hertwig’s epithelial root sheath cells are detectable on the surface of the root throughout root formation and do not disappear.33 Most of the Hertwig’s epithelial root sheath cells are attached to the surface of the cementum, and others separate to become the epithelial rest of Malassez. Thus, we used the PDL cells surrounding the apical 1/3 and apical pulp cells, which are assumed to comprise all developing apical complexes, for studying hyperactive root formation in OFCD patients. We found that BCOR mutations did not affect the expression of the mesenchymal and epithelial cell markers VIM and KRT14, respectively, in our cultured cells. Mesenchymal and epithelial markers were more highly expressed in cultured PDL cells than in pulp culture. Moreover, cultured PDL cells also showed higher BCOR expression than pulp cells, whether wild-type or mutant. It was previously reported that BCOR expressed in the mesenchyme has an important role in proper tooth formation.15 Therefore, our results suggested that BCOR is highly expressed in the mesenchyme and that our cultured PDL expressed BCOR at higher levels than pulp cells because of the predominance of mesenchymal cells in the PDL cultures. Compared with wild-type, BCOR expression was normal in pulp but reduced in PDL cells, which was consistent with our finding of higher rates of NMD in PDL. Moreover, in PDL but not pulp cell cultures, the mutant PDL had unstable mutant transcripts and proliferated faster than wild-type cells. This evidence suggests that the mutant PDL cells have insufficient BCOR function to repress target genes, resulting in the promotion of cell proliferation that contributes to hyperactive root formation.
In a previous report, mutant BCOR transcript levels did not significantly differ from normal, and mutant MSCs proliferated faster than control cells.2 That study used MSCs from the root apical papilla of an OFCD patient, and these cells may have different phenotypes than the differentiated pulp or PDL cells used in the present study, although our PDL cells were also harvested from the apical 1/3 and may have included apical papilla cells. Further studies are needed. However, the current study revealed that PDL cells might be more involved than pulp cells in the pathogenesis of radiculomegaly resulting from BCOR mutations. The mechanism and function will be studied further.
Conclusion
The nonsense mutation in patient 1 and the frameshift mutation in patient 2 were found to contribute to OFCD syndrome. A variation in phenotype severity was observed, and thus, further studies of factors such as X-chromosome inactivation or epigenetic modifications are needed to clarify the genotype–phenotype relationship. In patient 2, the mutation leading to the PTC-induced NMD mechanism in PDL cells caused unstable mutant transcripts and increased cell proliferation, which may be involved in hyperactive root formation.
Accession codes
References
Hedera, P. & Gorski, J. L. Oculo-facio-cardio-dental syndrome: skewed X chromosome inactivation in mother and daugther suggest X-linked dominant inheritance. Am. J. Med. Genet. A. 123A, 261–266 (2003).
Fan, Z., Yamaza, T., Lee, J. S., Yu, J., Wang, S., Fan, G. et al. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat. Cell. Biol. 11, 1002–1009 (2009).
Ng, D., Thakker, N., Corcoran, C. M., Donnai, D., Perveen, R., Schneider, A. et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat. Genet. 36, 411–416 (2004).
Davoody, A., Chen, I.-P., Nanda, R., Uribe, F. & Reichenberger, E. J. Oculofaciocardiodental (OFCD) syndrome: a rare case and review of the literature. Cleft Palate Craniofac. J. 49, e55–e60 (2012).
Hilton, E., Johnston, J., Whalen, S., Okamoto, N., Hatsukawa, Y., Nishio, J. et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. Eur. J. Hum. Genet. 17, 1325–1335 (2009).
Oberoi, S., Winder, A. E., Johnston, J., Vargervik, K. & Slavotinek, A. M. Case reports of oculofaciocardiodental syndrome with unusual dental findings. Am. J. Med. Genet. A. 136, 275–277 (2005).
Horn, D., Chyrek, M., Kleier, S., Luttgen, S., Bolz, H., Hinkel, G. K. et al. Novel mutations in BCOR in three patients with oculo-facio-cardio-dental syndrome, but none in Lenz microphthalmia syndrome. Eur. J. Hum. Genet. 13, 563–569 (2005).
Schulze, B. R., Horn, D., Kobelt, A., Tariverdian, G. & Stellzig, A. Rare dental abnormalities seen in oculo-facio-cardio-dental (OFCD) syndrome: three new cases and review of nine patients. Am. J. Med. Genet. 82, 429–435 (1999).
Huynh, K. D., Fischle, W., Verdin, E. & Bardwell, V. J. BCoR, a novel co-repressor involved in BCL-6 repression. Genes Dev. 14, 1810–1823 (2000).
Forrester, S., Kovach, M. J., Reynolds, N. M., Urban, R. & Kimonis, V. Manifestations in four males with and an obligate carrier of the Lenz microphthalmia syndrome. Am. J. Med. Genet. 98, 92–100 (2001).
Ng, D., Hadley, D. W., Tifft, C. J. & Biesecker, L. G. Genetic heterogeneity of syndromic X-linked recessive microphthalmia-anophthalmia: is Lenz microphthalmia a single disorder? Am. J. Med. Genet. 110, 308–314 (2002).
Wamstad, J. A. & Bardwell, V. J. Characterization of Bcor expression in mouse development. Gene Expr. Patterns 7, 550–557 (2007).
Hilton, E. N., Manson, F. D., Urquhart, J. E., Johnston, J. J., Stavotinek, A. M., Hedera, P. et al. Left-sided embryonic expression of the BCL-6 corepressor, BCOR, is required for vertebrate laterality determination. Hum. Mol. Genet. 16, 1773–1782 (2007).
Wamstad, J. A., Corcoran, C. M., Keating, A. M. & Bardwell, V. J. Role of the transcriptional corepressor Bcor in embryonic stem cell differentiation and early embryonic development. PLoS ONE 3, e2814 (2008).
Cai, J., Kwak, S., Lee, J.-M., Kim, E.-J., Lee, M.-J., Park, G.-H. et al. Function analysis of mesenchymal Bcor in tooth develoment by using RNA interference. Cell. Tissue. Res. 341, 251–258 (2010).
Linde, L., Boelz, S., Neu-Yilik, G., Kulozik, A. E. & Kerem, B. The efficiency of nonsense-mediated mRNA decay is an inherent character and varies among different cells. Eur. J. Hum. Genet. 15, 1156–1162 (2007).
Dedman, A. M., Majeed, Y., Tumova, S., Zeng, F., Kumar, B., Munsch, C. et al. TRPC1 transcript variants, inefficient nonsense-mediated decay and low up-frameshift-1 in vascular smooth muscle cells. BMC. Mol. Biol. 12, 30 (2011).
Holbrook, J. A., Neu-Yilik, G., Hentze, M. W. & Kulozik, A. E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 36, 801–808 (2004).
Tsukawaki, H., Tsuji, M., Kawamoto, T. & Ohyama, K. Three cases of oculo-facio-cardio-dental (OFCD) syndrome. Cleft Palate Craniofac. J. 42, 467–476 (2005).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Song, J. S., Hwang, D. H., Kim, S.-O., Jeon, M., Choi, B.-J., Jung, H.-S. et al. Comparative gene expression analysis of the human periodontal ligament in deciduous and permanent teeth. PLoS ONE 8, e61231 (2013).
Huang, A H-C, Chen, Y.-K., Lin, L.-M., Shieh, T.-Y. & Chan, A W-S Isolation and characterization of dental pulp stem cells from a supernumerary tooth. J. Oral. Pathol. Med. 37, 571–574 (2008).
Chen, Y. K., Huang, A. H., Chan, A. W. & Lin, L. M. Human dental pulp stem cells derived from cryopreserved dental pulp tissues of vital extracted teeth with disease demonstrate hepatic-like differentiation. J. Tissue. Eng. Regen. Med. (2013).
Carter, M. S., Doskow, J., Morris, P., Li, S., Nhim, R. P., Sandstedt, S. et al. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J. Biol. Chem. 270, 28995–29003 (1995).
Nelson, S. J. & Ash, M. M. Wheeler's Dental Anatomy, Physiology and Occlusion 9 edn. (Saunders Elsevier: Missouri, USA, 2010).
Lozic, B., Ljubkovic, J., Panduric, D. G., Saltvig, I., Kutsche, K., Krzelj, V. et al. Oculo-facio-cardio-dental syndrome in three succeeding generations: genotypic data and phenotypic features. Braz. J. Med. Biol. Res. 45, 1315–1319 (2012).
Frischmeyer, P. A. & Dietz, H. C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 8, 1893–1900 (1999).
Zhang, J., Sun, X., Qian, Y. & Maquat, L. E. Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-RNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA 4, 801–815 (1998).
Zhang, J., Sun, X., Qian, Y., LaDuca, J. P. & Maquat, L. E. At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: possible link between nuclear splicing and cytoplasmic translation. Mol. Cell. Biol. 18, 5272–5283 (1998).
Huang, X., Bringas, P. Jr, Slavkin, H. C. & Chai, Y. Fate of HERS during tooth root development. Dev. Biol. 334, 22–30 (2009).
Xu, L., Tang, L., Jin, F., Liu, X. H., Yu, J. H., Wu, J. J. et al. The apical region of developing tooth root constitutes a complex and maintains the ability to generate root and periodontium-like tissues. J. Periodontal Res. 44, 275–282 (2009).
Fang, J., Tang, L., Liu, X. H., Wen, L. Y. & Jin, Y. Changes of the unique odontogenic properties of rat apical bud cells under the developing apical complex microenvironment. Int. J. Oral Sci. 1, 26–33 (2009).
Huang, X. F. & Chai, Y. Molecular regulatory mechanism of tooth root development. Int. J. Oral Sci. 4, 177–181 (2012).
Acknowledgements
This work was supported by a Grant-in-Aid for young scientists (B); 22792038 from JSPS and the GCOE program at Tokyo Medical and Dental University. We would like to thank Dr Kerstin Kutsche, Universitatsklinikum Hamburg-Eppendorf, Hamburg, Germany, and Dr Hitoyata Shimokawa, Tokyo Medical and Dental University, Tokyo, Japan, for their kind advice. We also thank all patients and appreciate their cooperation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Surapornsawasd, T., Ogawa, T., Tsuji, M. et al. Oculofaciocardiodental syndrome: novel BCOR mutations and expression in dental cells. J Hum Genet 59, 314–320 (2014). https://doi.org/10.1038/jhg.2014.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.24
Keywords
This article is cited by
-
A novel deletion mutation in the BCOR gene is associated with oculo-facio-cardio-dental syndrome: a case report
BMC Pediatrics (2022)
-
BCOR mutations and unstoppable root growth: a commentary on oculofaciocardiodental syndrome: novel BCOR mutations and expression in dental cells
Journal of Human Genetics (2014)
