
Abstract
Noonan syndrome (NS), an autosomal dominant multisystem disorder, is caused by the dysregulation of the RAS-MAPK pathway and is characterized by short stature, heart defects, pectus excavatum, webbed neck, learning problems, cryptorchidism and facial dysmorphism. We here present the clinical and molecular characterization of a family with NS and multiple giant cell lesions (MGCLs). The proband is a 12-year-old girl with NS and MGCL. Her mother shows typical NS without MGCL. Whole-exome sequencing of the girl, her mother and her healthy maternal grand parents revealed a previously unobserved mutation in exon 5 of the PTPN11 gene (c.598 A>T; p.N200Y), transmitted from the mother to the proband. As no other modification in the RAS-MAPK pathway genes as related to Rasopathies was detected in the proband, this report demonstrates for the first time that a unique mutation affecting this, otherwise unaffected signaling route, can cause both NS and NS/MGCL in the same family. This observation further confirms that NS/MGCL is not a distinct entity but rather that MGCL represents a rare complication of NS. Moreover, the localization of the p.N200Y mutation suggests an alternative molecular mechanism for the excessive phosphatase activity of the PTPN11-encoded protein.
Similar content being viewed by others


Introduction
Noonan syndrome (NS, MIM 163950) is an autosomal-dominant multisystem condition with variable clinical expression. It is characterized by short stature, heart defect, pectus deformity, short neck with webbing, cryptorchidism, developmental delay of variable degree and facial dysmorphism.1, 2, 3 A subset of NS patients have multiple giant cell lesions (MGCLs).4
NS (or closely related conditions) are caused by the dysregulation of the RAS-MAPK signaling pathway,3 where, 10 genes have been shown to be mutated—PTPN11 (∼in 50% of cases),5 SOS1 (13%), RAF1 (5–15%), RIT1 (9%),6 KRAS (<5%), NRAS (<1%), SHOC2 (1.5%), BRAF (2%), MEK1 (1%) and CBL (<1%)4—leading to altered, usually increased, signaling. PTPN11, also known as SHP2, has an essential role as modulator of multiple signaling pathways.7, 8 It is composed of two SH2 domains, a catalytic phosphotyrosine phosphatase domain (PTP) and a C-terminal tail.9 In NS, a vast majority of known PTPN11 mutations are localized in or around the interacting surfaces of the N-SH2 and PTP domains, perturbing the equilibrium between active and inactive states and causing constitutive or prolonged activation of the protein.5, 10, 11, 12, 13
Here we describe the second uncovered mutation in the C-SH2 domain of the PTPN11, as identified by family exome sequencing.
Case report
A three-generation family displaying NS or NS/MGCL in the last two generations was studied. The proband girl (NS/MGCL; patient 1), her mother (NS only; patient 2) and her healthy grand parents were analyzed. All subjects gave written informed consent to participate in the study. See Supplementary text for Methods.
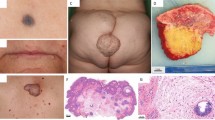
The proband (patient 1), a 12-year-old girl, is the third child of non-consanguineous parents. She showed mild psychomotor delay. Walking was acquired at 18 months. She also showed mild speech delay and slight learning difficulties. She was referred for evaluation of pain in the knees and ankles at 74/12 years of age. Clinical examination revealed short stature (110.8 cm; −2s.d.), moderate hemi-facial asymmetry, secondary to an expansive mandibular growth. She also had characteristic facial features of NS: high anterior, depressed nasal bridge, hypertelorism, down-slanting palpebral fissures and low-set and posteriorly angulated ears with thick helices (Figure 1). Radiolucent lesions in ascending rami of the right mandible were found. Partial bilateral resection was carried out at the age of 8 years, where pathology revealed the nature of lesions as MGCL (Figure 2). Progressive bilateral enlargement of pigmented villonodular synovitis (PVNS) lesions and recurrence of this tumor in ankles, knees and mandibule made further surgery mandatory. Treatment by Imatinib (Glivec) was initiated at the age of 116/12 years11, 12 and was rapidly effective in clinical and imaging evaluations of PVNS lesions. She had mild learning disabilities. There were no major cardiac abnormalities. No phenotypical specificity was observed as compared with previously reported cases (Supplementary Table 1). At the last evaluation, at 126/12 years of age, her weight, height and occipital-frontal circumference were 19.6 kg (−2.5s.d.), 122 cm (−3.5s.d.), 51.5 cm (−1.0s.d.), respectively. Patient 2 is the mother of patient 1. She had neonatal feeding difficulties, a congenital septal ventricular defect (spontaneously healed) and mild learning difficulties. She also had short stature (151 cm) and characteristic facial features of NS: hypertelorism, down-slanting palpebral fissures and low-set and posteriorly angulated ears with thick helices.

Clinical features of patient 1 at age of 8 years. (a) Note the short neck, ptosis, hypertelorism and down-slating palpebral fissures. (b) In the lateral view we see posteriorly low-set ears.

Biopsy of the left ankle (patient 1)—hematoxylin-eosin-saffron staining; original magnification × 200. Pigmented villonodular synovitis, an interesting thick synovial tissue with an abundant mononuclear cell component. Most of these cells are pigmented by amounts of hemosiderin. Some giant cells are observed (arrows) that are sometimes associated with foam or xanthomatous cells (not seen on the picture).
Exome sequencing
The exome of all four subjects (the proband, her mother and her grand-parents) were sequenced. On average 48 036 SNPs and 1667 indels were identified in each individual. For SNPs, we focused only on the 8563 protein altering events. For indels, only frameshift, non-frameshift, stoploss, stopgain and splicing events were taken into account, which corresponds to an average number of 167.
To identify the candidate gene, the initial list of 8788 protein altering variants of patient 1 (proband) was filtered by excluding variants of dbSNP (build 135), 1000 Genome data and an in-house exome database. From the resulting 145 private variants, only 80 were inherited from the mother (patient 2). Finally, only 1 out of these 80 turned out to be absent in the grand parents. This variant is the first uncovered missense mutation (c.598 A>T) localized in the 5th exon of PTPN11 causing a p.N200Y change in the C-SH2 domain. Sanger sequencing confirmed that the mutation is specific to patients 1 and 2 and absent in the healthy grand parents (Figure 3).

Electropherograms of the PTPN11 gene mutation c.598 A>T (p.N200Y). (a) Wild-type sequence of the grand mother. (b) Wild-type sequence of the grand father. (c) Mutated sequence of patient 2. (d) Mutated sequence of patient 1 (proband).
In order to try to explain the MGCL/PVNS phenotype of patient 1, specific variants of this subject were screened for the presence of genetic events modifying proteins of the RAS-MAPK pathway. Genes of the RAS-MAPK pathways were identified in recent reviews (Tartaglia and Gelb13 and Tidyman and Rauen14) or in dedicated databases (‘The ras-mapk syndromes HomePage’ or ‘NSEuroNet’). After narrowing the list of 8788 protein-altering variants of patient 1 by deleting modifications present in patient 2 and in the grand parents, a total of 404 events specific to patient 1 were identified (Supplementary Table 2). None of these were localized in the major genes of the RAS-MAPK pathway.
Discussion
A single, novel mutation in the PTPN11 was identified in the proband with NS/MGCL. It occurred de novo in her mother with NS only.
MGCLs have repeatedly been observed in NS patients, a condition initially named Noonan-like/MGCL syndrome,15, 16. Formerly considered as a separate entity, it is now generally admitted that MGCLs are part of the NS spectrum.4 We were able to show that no specific variant of patient 1 was localized in other Rasopathy-related genes of the RAS-MAPK pathway indicating that a single mutation affecting this signaling pathway can cause both NS and NS/MGCL, which is consistent with recent findings suggesting that MGCL may be a transitory NS feature that could resolve after puberty.
Most of the NS-causing mutations affect N-SH2 residues and are localized in or close to the PTP-interacting surface. p.N200Y is only the second reported mutation in the C-SH2 domain. It has recently been proposed that following catalysis, the C-SH2 domain facilitates the switching of the protein from the opened (active), back to the closed (inactive) state; p.N200Y may thus have a negative impact on this facilitating function, as confirmed by predictive values of Polyphen2, Mutation Taster and SIFT (categorized as ‘possibly damaging’, ‘damaging’ and ‘disease causing’, respectively), although further biochemical/structural studies are needed to fully characterize the impact of this modification.
References
Mendez, H. M. & Opitz, J. M. Noonan syndrome: a review. Am. J. Med. Genet. 21, 493–506 (1985).
Noonan, J. A. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease. Am. J. Dis. Child. 116, 373–380 (1968).
Roberts, A. E., Allanson, J. E., Tartaglia, M. & Gelb, B. D. Noonan syndrome. Lancet 381, 333–342 (2013).
Tartaglia, M., Zampino, G. & Gelb, B. D. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol. Syndromol. 1, 2–26 (2010).
Tartaglia, M., Mehler, E. L., Goldberg, R., Zampino, G., Brunner, H. G., Kremer, H. et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 29, 465–468 (2001).
Aoki, Y., Niihori, T., Banjo, T., Okamoto, N., Mizuno, S., Kurosawa, K. et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am. J. Hum. Genet. 93, 173–180 (2013).
Feng, G. S. Shp-2 tyrosine phosphatase: Signaling one cell or many. Exp. Cell. Res. 253, 47–54 (1999).
Stein-Gerlach, M., Wallasch, C. & Ullrich, A. SHP-2, SH2-containing protein tyrosine phosphatase-2. Int. J. Biochem. Cell B. 30, 559–566 (1998).
Neel, B. G., Gu, H. & Pao, L. The 'Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends. Biochem. Sci. 28, 284–293 (2003).
Tartaglia, M., Kalidas, K., Shaw, A., Song, X., Musat, D. L., van der Burgt, I. et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 70, 1555–1563 (2002).
Blay, J. Y., El Sayadi, H., Thiesse, P., Garret, J. & Ray-Coquard, I. Complete response to imatinib in relapsing pigmented villonodular synovitis/tenosynovial giant cell tumor (PVNS/TGCT). Ann. Oncol. 19, 821–822 (2008).
Cassier, P. A., Gelderblom, H., Stacchiotti, S., Thomas, D., Maki, R. G., Kroep, J. R. et al. Efficacy of imatinib mesylate for the treatment of locally advanced and/or metastatic tenosynovial giant cell tumor/pigmented villonodular synovitis. Cancer 118, 1649–1655 (2012).
Tartaglia, M. & Gelb, B. D. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 1214, 99–121 (2010).
Tidyman, W. E. & Rauen, K. A. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19, 230–236 (2009).
Cohen, M. M. Jr. & Gorlin, R. J. Noonan-like/multiple giant cell lesion syndrome. Am. J. Med. Genet. 40, 159–166 (1991).
Beneteau, C., Cave, H., Moncla, A., Dorison, N., Munnich, A., Verloes, A. et al. SOS1 and PTPN11 mutations in five cases of Noonan syndrome with multiple giant cell lesions. Eur. J. Hum. Genet. 17, 1216–1221 (2009).
Acknowledgements
This work was supported by funds from the Strasbourg High Throughput Next Generation Sequencing facility (GENOMAX) and INSERM UMR_S1109.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Carapito, R., Paul, N., Untrau, M. et al. A new mutation in the C-SH2 domain of PTPN11 causes Noonan syndrome with multiple giant cell lesions. J Hum Genet 59, 57–59 (2014). https://doi.org/10.1038/jhg.2013.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.118
