
Abstract
Late-onset multiple carboxylase deficiency, also known as biotinidase (BTD) deficiency, is an autosomal recessively inherited disorder of biotin metabolism. Its early diagnosis and treatment seems that it can even fully prevent its various clinical manifestations. Mutations in the BTD gene scattered throughout its coding region have been detected in patients ascertained either through newborn screening or clinically. From March 2010 up to June 2011, 18 954 Greek neonates were subjected to biochemical determination of BTD activity through a semiquantitative fluoroimmunoassay. Subsequently, the first cohort of our ‘suspected’ samples was further tested for the presence of aberrations associated either with partial or profound BTD deficiency through sequencing of the coding region of the BTD gene, including splice-site junctions. On the basis of the molecular data derived from the study of our first cohort of ‘suspected’ samples, a panel of four mutations, most frequently encountered in the Greek population, was created, and a rapid, reliable and cost-effective real-time-based genotyping assay for the detection of these mutations was developed. This is the first report about the BTD mutational spectrum in Greece, and it could be a beneficial utility in the differential clinical diagnosis of BTD deficiency.
Similar content being viewed by others

Introduction
Multiple carboxylase deficiency (MCD) is a disorder of biotin metabolism, inherited with autosomal recessive mode. It is attributed either to mutations occurring in the BTD (biotinidase) gene resulting in the manifestation of the late-onset MCD or to mutations in the HLCS (holocarboxylase synthetase) gene giving rise to the early-onset MCD syndrome (OMIM number 253260). As the emergence of clinical symptoms, some of which are associated with severe impairment of the central nervous system, may be even fully prevented through early biotin supplementation in both types of MCD,1 its early diagnosis is considered beneficial. For that reason, the determination of BTD activity has been incorporated in the routine newborn screening program of many countries.
Late-onset MCD, also known as BTD deficiency syndrome, is distinguished to partial and profound type on the basis of the mean enzyme–serum activity observed. Particularly, 10–30% of mean BTD serum activity is implicated to partial enzyme deficiency, whereas <10% activity to profound enzyme deficiency. The aforementioned biochemical data have been correlated with the presence of mutations in the BTD gene through genetic testing of positive samples.2 Specific mutations, such as the frequent p.Asp444His mutation, either in homozygosity or combined with other mutations, labeled as ‘severe’, are present in clinical cases of partial deficiency, whereas the ‘severe’ mutations either in homozygosity or in compound heterozygosity are implicated in the profound form of BTD deficiency.3 The carrier frequency of BTD mutations is approximately estimated to 1 in 120, whereas the combined incidence of profound and partial cases are about 1 in 60 000, worldwide.2, 4
Up to date, more than 100 mutations of the BTD gene have been recorded. These mutations are scattered throughout the coding region of the BTD gene and can be of any type.1 Two distinct mutations in compound heterozygosity are detected in most clinical cases, except in populations characterized by significant consanguinity.5
The phenotype of the disorder includes a variety of clinical symptoms such as neurological (seizures, ataxia, hypotonia, developmental delay, hearing loss, problematic vision), gastroenterological (vomiting, feeding difficulties), dermatological (alopecia, skin rash) and even respiratory ones (apnea, tachypnea).2, 6 The aforementioned heterogeneity regarding the clinical manifestations of the late-onset MCD syndrome could be explained by the diverse functions of the BTD protein. In fact, BTD has a critical role in the recycling of both free- and bound biotin, which is essential for the processing of amino acids, lipid and carbohydrate metabolic pathways. Furthermore, there is emerging evidence that BTD mediates both biotinylation and debiotynilation of histones, and consequently affecting processes such as DNA repair, replication and transcription.7, 8, 9, 10, 11 It is worth noting that the clinical manifestations of patients carrying the same mutations may vary even between members of the same family concerning the severity, as well as the age of onset. This might be due to the simultaneous presence of rare variants and/or epigenetic modifications, the action of modifier loci or even environmental effects.1
The aim of this study was to successfully identify the ‘suspected’ samples under routine newborn screening procedures and to report the BTD mutational spectrum in the Greek population. More specifically, the frequency and the type of pathogenic alterations encountered in patients ascertained through newborn screening have been elucidated, and a rapid DNA-based confirmatory protocol for the ‘suspected’ samples has been developed.
Materials and methods
Patients
Blood samples were collected from neonates on Whatman filter papers following the standard procedure, to be screened for the presence of a series of metabolic disorders, as well as cystic fibrosis, congenital adrenal hyperplasia and BTD deficiency. The newborn screening procedure, including confirmatory genetic testing was approved by the Institutional Review Board of IASO hospital, in agreement with the 1975 Helsinki statement, revised in 1983. A coded designation was utilized to protect patient anonymity.
Determination of enzyme activity
A semiquantitative fluoroimmunoassay (Delfia Biotinidase kit; Perkin Elmer, Boston, MA, USA) was adopted to determine the BTD activity on dried blood spots. According to the manufacturer's instructions, values <19 nmol min−1 dl−1 are implicated to profound BTD deficiency, whereas values >49 nmol min −1dl−1 are considered as normal. The obtained values, which fell in the range between 19 nmol min−1 dl−1 and 49 nmol min−1dl−1, indicate possible partial enzyme deficiency.
DNA extraction from dried blood spots
Genomic DNA was extracted from dried blood spots following the standard chelex protocol (Chelex 100, Molecular Biology Grade Resin, Biorad, Athens, Greece). The quantity and quality of the DNA samples was determined with ‘NanoDrop’ (Thermo Fisher Scientific, Wilmington, MA, USA).
DNA sequencing
Exons 1–4 of the BTD gene as well as their flanking regions were amplified by PCR. The PCR products were purified with ‘Multiscreen PCRμ96 Filter Plate’ (Millipore, Malva, Athens, Greece), and then subjected to an automated cycle sequencing with the Big Dye Terminator Cycle v3.1 sequencing kit (Applied Biosystems, Foster City, CA, USA) and electrophoresis on an ABI Prism 310 Sequencer (Perkin Elmer, Applied Biosystems). Primer pairs were designed with NCBI/Primer Blast Tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) and are available upon request.
Real-time PCR-based genotyping assay
Four distinct real-time PCR assays were developed for the rapid screening of ‘suspected’ samples concerning the presence of p.Asp444His, p.Gln456His, p.Thr532Meth and c.98_104del7ins3 mutations. Primers and probes for these assays were synthesized by VBC Genomics Bioscience Research GmbH (Vienna, Austria). The total volume of the PCR reaction was 10 μl and consisted of 1 × KAPA PROBE FAST Universal 2 × qPCR Master Mix (Kappa Biosystems, Boston, MA, USA), 0.75 μM forward/reverse primers mix, 0.5 μM HEX/6FAM probes MIX and 3.75 ng μl−1 of DNA. All PCR reactions were performed in a Rotor-Gene 6000 (Corbett Research, Bioanalytica S.A., Athens, Greece). The cycling conditions were as follows: 60 °C for 30 s (1 cycle), 95 °C for 10 min (1 cycle), 95 °C for 15 s or 60 °C for 1 min (45 cycles), 60 °C for 30 s (1 cycle). Positive controls, either a heterozygote or a homozygote sample for each mutation, as well as non-template controls were included in each PCR run, so as to check reaction efficiency and to exclude any contaminants’ presence.
Results
Screening for BTD deficiency among neonates
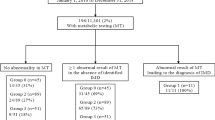
From March 2010 up to June 2011, 18 954 neonates born in Athens were subjected to massive biochemical determination of BTD activity through a semiquantitative fluoroimmunoassay. Values between 25 nmol min−1 dl−1 and 50 nmol min−1 dl−1 were obtained in 69 samples, which were further tested through molecular techniques to confirm the primary biochemical result.
Mutational analysis
The confirmatory molecular testing procedure of our study was conducted in two stages. During the first stage, 44 infants with values indicative of partial BTD deficiency were screened for the presence of point mutations or small insertions/deletions through sequencing of the coding region of the BTD gene, including splice site junctions. In the second stage, 25 ‘suspected’ samples were screened for the four mutations most frequently encountered in the cohort of the ‘suspected’ samples of the first stage, through a rapid real-time based PCR genotyping assay developed in our laboratory.
Regarding the first stage of our molecular study, 19 of the 44 neonates found to carry a mutation in one allele associated either with profound or partial BTD deficiency. Specifically, nine were carriers of the rather common p.Asp444His mutation, which is associated with 50% decreased enzyme activity. The p.Asp444His mutation, in combination with a ‘severe’ aberration, results in partial BTD deficiency, as it seems to be the case in 3 out of the 44 samples (Table 1). Moreover, one sample of our primary cohort carried the aforementioned mutation in both alleles. The particular genotype has been designated to partial loss of enzyme activity (∼25% of mean serum activity) on the basis of the literature.12, 13 The most common ‘severe’ alterations worldwide, p.Gln456His and p.Cys33PhefsTer36, were detected in 5 out of the 19 samples (p.Gln456His: BTD 2, 3 and 42; p.Cys33PhefsTer36: BTD 4 and 35).
Furthermore, a novel amino-acid change in codon 211, which resulted from the c.632G>A nucleotide change, was identified. Its effect on protein level has been evaluated through SIFT (http://sift.jcvi.org/www/SIFT_seq_submit2.html) and PolyPhen (http://genetics.bwh.harvard.edu/pph/) algorithms. According to the results of the above predictor tools, the p.Arg211His alteration is benign (Table 2). The Human Splice Finder (v.2.4) web tool (http://www.umd.be/HSF/) was also used to find out any potential effect of the particular nucleotide change (c.632G>A) on the splicing process. It is really interesting that the Human Splice Finder main algorithm implicates this nucleotide change in the abrogation of the splice donor site, as well as the fact that the other algorithms used for the prediction of enhancer/silencer motifs incorporated in the Human Splice Finder main page indicate that the c.632G>A not only induces the abrogation of enhancer-binding sites, but also the creation of a silencer motif (Table 2).
Finally, one nonsense, p.Gln88Ter, and two missense mutations, p.Val199Met and p.Thr532Met, have been identified in 4 out of the 19 carriers. Except from mutations, three silent polymorphisms were recorded, too (Table 3).
During the second stage of our study, 25 ‘suspected’ samples were subjected to mutational analysis. Five newborns carried in heterozygosity and one in homozygosity of the p.Asp444His mutation, whereas two others were carriers of the p.Thr532Met and p.Cys33PhefsTer36, respectively (Table 4). The seven carriers detected by our rapid molecular screening method were further subjected to sequencing of the whole coding region of the BTD gene, to exclude the possible presence of a second aberration. However, no other mutation was detected in any of these carriers. All the above data are summarized in Tables 1 and 4.
Discussion
BTD deficiency is a metabolic disorder with variable phenotypic expression, as it seems to affect multiple organic systems. False-positive results might occur in the biochemical assay (screening procedure) for various reasons, such as infant's prematurity or mishandling of the sample before its arrival in the laboratory. However, mutational analysis seems to be able to readily discriminate between patients heterozygote for profound BTD deficiency and patients suffering from partial enzyme deficiency.2, 4, 16 The aforementioned facts motivated our laboratory to incorporate genetic testing as a second-tier screening step in the cohort of the ‘suspected’ samples.
In total, eight distinct germ line mutations, p.Asp444His, p.Gln456His, p.Cys33PhefsTer36, p. Thr532Met, p.Arg157His, p.Val199Met, p.Cys186Tyr and p.Arg211His, were identified in our cohorts of ‘suspected’ samples. The p.Arg211His has never been described before to the best of our knowledge. On the basis of the results of the in-silico analysis, it seems that it rather affects the splicing process. However, this finding requires molecular confirmation through further study of the transcripts produced in vitro and in vivo by the particular variant. Clinical studies should also be conducted to establish or not, the putative pathogenicity of the particular variation.
Specifically, five cases of partial BTD deficiency were identified in a cohort of 18 954 newborns, based on the combined data of the biochemical and molecular assays employed in our laboratory. Therefore, the incidence of partial BTD deficiency seems to be rather high in the Greek population. Particularly, it is estimated to be approximately 1:3791 births, resembling the ones observed in Brazil and Turkey.5, 17 It is also worth noting the fact that a large proportion of our ‘positive’ samples (26 of 69) are found to be merely carriers of a pathogenic mutation associated either to profound or to partial BTD deficiency in both cohorts of our study. The potential of the molecular testing to differentiate the condition of partial deficiency from the carrier status is therefore confirmed.
As far as the frequency of mutations associated with partial or profound BTD deficiency is concerned, our results confirm the high frequency of the p.Asp444His allele, as it was encountered in carrier status, or combined with another pathogenic alteration or even in homozygosity at approximately 29% of the ‘suspected’ neonates subjected to genetic testing. Each of the common p.Gln456His and p.Cys33PhefsTer36 alleles, the former detected only among newborn screening groups and the latter detected mainly among clinically diagnosed patients,18 have been encountered in the 4.34% of our specimen. Nevertheless, the double mutation p.Ala171Thr:P.Asp444His and the aberration p.R538C, which are considered as common alleles among neonates in USA studies,1 were not detected in the Greek group of ‘suspected’ samples ascertained through newborn screening. Regarding the latter mutation, its absence in our cohort may be explained by the fact that its frequency seems to be significantly higher in the clinically ascertained group of patients rather than the one based on newborn screening biochemical data.18
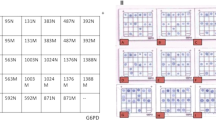
It is further worth noting that mutations described initially in the Turkish population such as the p.Thr532Met, and the p.Cys186Tyr,5 have been detected in our Greek cohort, too. Particularly, the p.Thr532Met, which seems to be a rather common allele in Turkey, was identified at about 2.9% of our selected samples. Subsequently, it has been included in the panel of the four mutations (p.Asp444His, p.Gln456His, p.Cys33PhefsTer36, p.Thr532Met) devised for initial screening of our second group of ‘suspected’ samples. The choice of the mutations included in this panel is based on the mutational spectrum of the BTD gene, as it has been shaped by the results of the first part of our molecular study (Figure 1).

Mutational spectrum of the BTD gene. This configuration represents schematically all the germline mutations associated with either types of BTD deficiency, which were detected in the specimen of our two-stage study. Novel alterations are indicated with italics.
Seven carriers of the p.Asp444His (5 of 25), p.Cys33PhefsTer36 (1 of 25) and p.Thr532Met (1 of 25), as well as one homozygote for the p.Asp444His were identified in our second cohort of 25 ‘suspected’ samples, which has been initially subjected to testing only for the presence of the panel mutations. Therefore, the high frequency of the selected mutations among specimens with values indicative of the BTD deficiency was further confirmed. The seven heterozygote specimens were further subjected to complete sequencing of the BTD gene and proved to be negative for the presence of any other pathogenic alteration. The results from sequencing analysis indicated the efficiency of our panel to detect the ‘true-positive’ samples among ‘suspected’ specimens that emerged from the routine Greek newborn screening program.19 Nevertheless, as the rather small sample size of our second cohort of putative ‘positive’ samples could be considered as a limitation for definitive conclusions, a further study on a larger cohort should be conducted.
It would be also interesting to apply the aforementioned panel in a Greek group of clinically diagnosed patients, to find out if there are any differences concerning the spectrum of mutations encountered in the cases ascertained through newborn screening and the ones identified on the basis of clinical manifestations, as it has been observed in other studies.1, 18
In conclusion, our results indicate that the mutational spectrum of the BTD gene in the Greek population, reported for the first time on the basis of the newborn screening data, resembles more the one observed in the Turkish population, compared with that of USA cohorts. For that reason, we have slightly modified the panel of the five mutations (p.Asp444His, p.Gln456His, p.Cys33PhefsTer36, p.Ala171Met:p.Asp444His, p.Arg538Cys) recommended for routine screening by substituting the p.Ala171Met:p.Asp444His and the p.Arg538Cys mutations with the p.Thr532Met aberration. Furthermore, we developed a high throughput real-time-based PCR genotyping assay concerning the detection of the mutations included in our panel. In this way, we achieved the development of a rapid and cost-effective second-tier molecular testing with high detection rate regarding ‘true-positive’ samples, which can be incorporated in the expanded neonatal screening program of the Greek population.
References
Pindolia, K., Jordan, M. & Wolf, B. Analysis of mutations causing biotinidase deficiency. Hum. Mutat. 31, 983–991 (2010).
Cowan, T. M., Blitzer, M. G. & Wolf, B. Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet. Med. 12, 464–470 (2010).
Hymes, J., Stanley, C. M. & Wolf, B. Mutations in BTD causing biotinidase deficiency. Hum. Mutat. 18, 375–381 (2001).
Wolf, B. Worldwide survey of neonatal screening for biotinidase deficiency. J. Inherit. Metab. Dis. 14, 923–927 (1991).
Pomponio, R. J., Coskun, T., Demirkol, M., Tokatli, A., Ozalp, I., Huner, G. et al. Novel mutations cause biotinidase deficiency in Turkish children. J. Inherit. Metab. Dis. 23, 120–128 (2000).
Wolf, B., Grier, R. E., Allen, R. J., Goodman, S. I., Kien, C. L., Parker, W. D. et al. Phenotypic variation in biotinidase deficiency. J. Pediatr. 103, 233–237 (1983).
Gravel, R. A. & Narang, M. A. Molecular genetics of biotin metabolism: old vitamin, new science. J. Nutr. Biochem. 16, 428–431 (2005).
Hassan, Y. I. & Zempleni, J. Epigenetic regulation of chromatin structure and gene function by biotin. J. Nutr. 136, 1763–1765 (2006).
Kothapalli, N., Camporeale, G., Kueh, A., Chew, Y. C., Oommen, A. M., Griffin, J. B. et al. Biological functions of biotinylated histones. J. Nutr. Biochem. 16, 446–448 (2005).
Wolf, B. Biotinidase: its role in biotinidase deficiency and biotin metabolism. J. Nutr. Biochem. 16, 441–445 (2005).
Zempleni, J., Hassan, Y. I. & Wijeratne, S. S. Biotin and biotinidase deficiency. Expert Rev. Endocrinol. Metab. 3, 715–724 (2008).
Muhl, A., Moslinger, D., Item, C. B. & Stockler-Ipsiroglu, S. Molecular characterisation of 34 patients with biotinidase deficiency ascertained by newborn screening and family investigation. Eur. J. Hum. Genet. 9, 237–243 (2001).
Swango, K. L., Demirkol, M., Huner, G., Pronicka, E., Sykut-Cegielska, J., Schulze, A. et al. Partial biotinidase deficiency is usually due to the D444H mutation in the biotinidase gene. Hum. Genet. 102, 571–575 (1998).
Wang, J., Smith, P. J., Krainer, A. R. & Zhang, M. Q. Distribution of SR protein exonic splicing enhancer motifs in human protein-coding genes. Nucleic Acids Res. 33, 5053–5062 (2005).
Sironi, M., Menozzi, G., Riva, L., Cagliani, R., Comi, G. P ., Bresolin, N. et al. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 32, 1783–1791 (2004).
Wolf, B. Clinical issues and frequent questions about biotinidase deficiency. Mol. Genet. Metab. 100, 6–13 (2010).
Neto, E. C., Schulte, J., Rubim, R., Lewis, E., DeMari, J., Castilhos, C. et al. Newborn screening for biotinidase deficiency in Brazil: biochemical and molecular characterizations. Braz. J. Med. Biol. Res. 37, 295–299 (2004).
Norrgard, K. J., Pomponio, R. J, Hymes, J. & Wolf, B. Mutations causing profound biotinidase deficiency in children ascertained by newborn screening in the United States occur at different frequencies than in symptomatic children. Pediatr. Res. 46, 20–27 (1999).
Loukas, Y. L., Soumelas, G. S., Dotsikas, Y., Georgiou, V., Molou, E., Thodi, G. et al. Expanded newborn screening in Greece: 30 months of experience. J. Inherit. Metab. Dis. (in press).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Thodi, G., Molou, E., Georgiou, V. et al. Mutational analysis for biotinidase deficiency of a Greek patients’ cohort ascertained through expanded newborn screening. J Hum Genet 56, 861–865 (2011). https://doi.org/10.1038/jhg.2011.119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2011.119
Keywords
This article is cited by
-
The novel homozygous p.Asn197_Ser201del mutation in BTD gene is associated with profound biotinidase deficiency in an Iranian consanguineous family
Molecular Biology Reports (2020)
-
Clinical utility gene card for: Biotinidase deficiency—update 2015
European Journal of Human Genetics (2016)
-
Clinical utility gene card for: Biotinidase deficiency
European Journal of Human Genetics (2012)
