
Abstract
We have recently found that multiple rare variants of the glucocerebrosidase gene (GBA) confer a robust risk for Parkinson disease, supporting the ‘common disease-multiple rare variants’ hypothesis. To develop an efficient method of identifying rare variants in a large number of samples, we applied multiplexed resequencing using a next-generation sequencer to identification of rare variants of GBA. Sixteen sets of pooled DNAs from six pooled DNA samples were prepared. Each set of pooled DNAs was subjected to polymerase chain reaction to amplify the target gene (GBA) covering 6.5 kb, pooled into one tube with barcode indexing, and then subjected to extensive sequence analysis using the SOLiD System. Individual samples were also subjected to direct nucleotide sequence analysis. With the optimization of data processing, we were able to extract all the variants from 96 samples with acceptable rates of false-positive single-nucleotide variants.
Similar content being viewed by others
Introduction
Genome-wide association studies using common single-nucleotide polymorphisms have been conducted to explore disease susceptibility genes on the basis of the ‘common disease-common variants’ hypothesis. On the basis of these strategies, a number of genes across genomes with statistically significant association with diseases have successfully been identified.1 These studies have substantially contributed to our understanding of diverse molecular pathways underlying diseases. However, established susceptibility genes remain insufficient to understand the entire disease pathways and their odds ratios are generally small, not exceeding 2.0 in most cases, which may largely reflect the limitation of the common disease-common variants hypothesis. Thus, we are far from fully understanding the pathophysiology of diseases. The limitation is probably explained by the fact that genome-wide association studies using common single-nucleotide polymorphisms cannot provide information on rare variants.2
We and other groups have recently found that multiple rare variants of the glucocerebrosidase gene (GBA) confer a robust risk for Parkinson disease (PD).3, 4, 5, 6 Our findings have confirmed that multiple rare variants, which can be identified only by extensive resequencing, considerably contribute to the pathogenesis of common diseases such as PD.5, 6 However, even for a particular gene, it is tremendously laborious to detect all the variants in a large number of samples, and efficient methods of detecting multiple rare variants should be established.
Recently, new technologies using high-throughput sequencing platforms have been developed.7 To identify rare variants in a large number of samples, Sanger sequencing of individual samples would be accurate but remained laborious and expensive. Thus, improved methods would be required. To take advantage of high-throughput sequencing, technologies of barcode indexing8 and physical separation (segmentation) of samples on a slide have been introduced to further increase the throughput of sequencing. Although analysis of pooled DNAs would be a potentially robust approach, the accuracy/error rate of reads would be critical for determining the frequency of variants in pooled DNAs. Next-generation sequencers have a great advantage for obtaining an extraordinarily high throughput, but the accuracy of reads has been a technical issue. In particular, error rate can be specifically critical for analyzing pooled DNAs, because sequence analysis of pooled DNAs derived from a large number of samples may lead to erroneous detection of ‘variants.’
To overcome this problem, we need to determine the optimal conditions of pooling. Furthermore, barcode indexing of pooled DNAs should allow further multiplexing. As a model system, we applied multiplexed resequencing of six pooled DNA samples with 16 sets for barcode indexing using the SOLiD System (Life Technologies, Carlsbad, CA, USA) to evaluate the efficiency of this system for identifying multiple rare variants of GBA in 96 patients with PD.
Materials and methods
Sample DNA preparation and quantification
The participants were 96 patients with PD. Genomic DNA samples were extracted in accordance with the standard procedure from whole peripheral blood collected in tubes containing anticoagulant EDTA. After the extracted DNA samples were stored at 4 °C in TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0), their purity and concentration were determined using DU 730 Life Science (Beckman Coulter, CA, USA). Only DNA samples with an OD260/OD280 ratio of 1.8±0.1 were used. On the basis of the spectrophotometer reading of the stocks, TE buffer was added to DNA samples extracted from blood to obtain a target concentration of 100.0 ng μl−1 and was equilibrated at 4 °C for 24 h. Each 100.0 ng μl−1 sample was quantified using Spectramax Gemini XS (Molecular Devices, CA, USA), with a Quant-iT Picogreen dsDNA Assay kit (Life Technologies, CA, USA), and only samples with a concentration of 100.0±5.0 ng μl−1 were used for pooling. Samples whose concentration was above this were rediluted to 100.0 ng μl−1 on the basis of new readings; this process was repeated iteratively until a concentration of 100.0±5.0 ng μl−1 was obtained. Samples whose concentration was below this were rediluted from stocks through the same stages as above. This study was approved by the Ethical Committee of the University of Tokyo.
Preparation of pooled DNAs
Pooled DNAs were then prepared by combining equal volumes (5.0 μl) of each sample. Six samples were pooled as 16 sets of pooled DNAs.
Polymerase chain reaction amplification
Each set of pooled DNAs was subjected to polymerase chain reaction (PCR) using TaKaRa LA Taq (Takara, Shiga, Japan) to amplify the target gene (GBA).
Three primer pairs were used to selectively amplify all the exons and respective flanking intronic regions of GBA but not its pseudogene, as described earlier.9 Primers for exons 1–5 were 5′-CCTAAAGTTGTCACCCATAC-3′ and 5′-ACCTACCCTACAGTTT-3′, those for exons 5–7 were 5′-GACCTCAAATGATATACCTG-3′ and 5′-AGTTTGGGAGCCAGTCATTT-3′ and those for exons 8–11 were 5′-TGTGTGCAAGGTCCAGGATCAG-3′ and 5′-ACCACCTAGAGGGGAAAGTG-3′. The sizes of amplicons were 2953 base pairs (bp), 2026 and 1670 bp, respectively. PCR was performed in 50 μl reaction volumes containing 5.0 μl of 10 × LA PCR Buffer II (Mg2+-free), 5.0 μl of 25 mM MgCl2, 8.0 μl of dNTP mixture (2.5 mM each), 0.4 μM of each primer, 50 ng of pooled DNAs and 2.5 U TaKaRa LA Taq DNA polymerase. PCR was performed in a PTC-200 DNA Tetrad thermal cycler (Life Science Research, CA, USA) as follows: 94 °C for 5 min, followed by 5 cycles of 94 °C for 20 s, 62 °C for 30 s and 68 °C for 3 min; 5 cycles of 94 °C for 20 s, 60 °C for 30 s and 68 °C for 3 min and 25 cycles of 94 °C for 20 s, 58 °C for 30 s and 68 °C for 3 min. Final extension was completed at 68 °C for 7 min. Then, 50 μl of the PCR product was subjected to 1% agarose gel electrophoresis (125 V for 30 min), followed by staining with ethidium bromide for 30 min. The DNA band corresponding to the PCR product (2953 bp for exons 1–5, 2026 bp for exons 5–7 and 1670 bp for exons 8–11) visualized on a UV transilluminator was excised with a clean razor blade. The DNA fragment was recovered from excised agarose gel using a GenElute Agarose Spin Column (Sigma, MO, USA).
Each amplicon was quantified using Spectramax Gemini XS, with a Quant-iT Picogreen dsDNA Assay kit. The final amount of PCR products used for subsequent library preparations were 2.95 μg for exons 1–5 amplicons, 2.02 μg for exons 5–7 amplicons and 1.67 μg for exons 8–11 amplicons. Samples whose amount was below this were repeatedly subjected to PCR amplifications using the same procedures as above. In each set of pooled DNAs, three amplicons were pooled into one tube.
Preparation of sequencing library
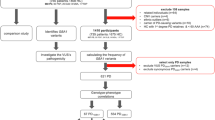
Sequencing libraries were prepared as follows. The pooled amplicons containing 6.64 μg of PCR products (2.95 μg for exons 1–5 amplicons, 2.02 μg for exons 5–7 amplicons and 1.67 μg for exons 8–11 amplicons) were subjected to fragmentation. Fragmentation was achieved using the Covaris S2 System (Covaris, MA, USA) under the following conditions: number of cycles, 10; bath temperature, 5 °C; duty cycle, 20%; intensity, 10; cycles per burst, 1000 and time, 60 s, which were optimal for the generation of 50–110 bp fragments. Fragmentation was followed by repair of DNA ends using an End-It DNA end-repair kit (Epicentre Biotechnologies, WI, USA), ligation of adaptors with barcode indexing using Quick ligase enzyme (New England BioLabs, MA, USA) and nick translation using DNA polymerase I (New England BioLabs). Fragmented DNAs of 130–190 bp were fractionated by 4% agarose gel electrophoresis. Fragmented DNAs from agarose gel were eluted into 20 μl volume using a MinElute Gel Extraction kit (Qiagen, CA, USA). Then, PCR amplification of the fragmented DNAs was performed in 52 μl reaction volumes containing 50 μl of Invitrogen Platinum PCR SuperMix (Life Technologies), 2 μM of each PCR primer and 2 μl of fragmented DNAs. PCR was performed in a PTC-200 DNA Tetrad thermal cycler (Life Science Research) as follows: 95 °C for 5 min, followed by 12 cycles of 95 °C for 15 s, 62 °C for 15 s and 70 °C for 1 min. Final extension was completed at 70 °C for 5 min. Fragmented DNAs containing 60–110 bp genomic DNA inserts excluding adaptor sequences were fractionated by 4% agarose gel electrophoresis. Fragmented DNAs from agarose gel were eluted into 20 μl volumes using a MinElute Gel Extraction kit (Qiagen). The prepared sequence libraries were then subjected to resequencing analysis using the SOLiD System (Life Technologies) to generate sequence reads of 50 bp. The workflow of procedures is shown in Figure 1.

Workflow of preparation of sets of pooled DNAs and generation of sequence library. Sixteen sets of pooled DNAs were prepared: pooling of six samples. Dots represent individual DNA samples and tubes represent sets of pooled DNAs. Each set of pooled DNAs was subjected to amplification of the target gene (GBA), and the sequence library for the SOLiD System was then prepared (described in square frames). Barcode-indexed samples were pooled into one tube.
Barcode indexing
We used six-base barcode indexing in accordance with the manufacturer's instructions. Sixteen different adaptors were used and each adaptor contains a different barcode (5′-GGGCTT-3′, 5′-GGTGTG-3′, 5′-AAGGGG-3′, 5′-CCGATG-3′, 5′-CAACGA-3′, 5′-GTGCCC-3′, 5′-GTCTGG-3′, 5′-ACGGAG-3′, 5′-GAAGGG-3′, 5′-GACCGC-3′, 5′-CTCAGG-3′, 5′-AGCGTT-3′, 5′-CGGGTC-3′, 5′-CGTCTG-3′, 5′-TAGCGT-3′ and 5′-GCGTTT-3′). For multiplexing, 16 different barcode-indexed libraries of pooled DNAs were pooled into one tube. After resequencing of target DNA, additional rounds of sequencing of barcode adaptors were performed and all reads were sorted by barcode.
Validation of variants
To evaluate the results, individual samples were also subjected to direct nucleotide sequence analysis to detect each variation within the amplicons. Amplifications were conducted using the same pairs of primers mentioned earlier. Primers for direct nucleotide sequence analysis were designed to cover the full length of amplicons (available on request).
Data analysis
Obtained sequence reads were sorted by barcode, and sorted reads were aligned to the reference sequences (6546 bp) masking repetitive sequences with filters that allow a maximum of two color space mismatches using Maq.10 To determine the error rate, the sequence reads of bases without variants in any of the pooled DNAs as confirmed by Sanger sequencing of individual samples were evaluated. The error rate is the ratio of the number of mismatched bases to that of the total aligned reads at each nucleotide position of reference sequences. For bases with variants in some samples of pooled DNAs that were confirmed by individual Sanger sequencing, we also calculated the fraction of variant reads using the ratio of the number of mismatched reads to the total number of aligned reads at each nucleotide position of reference sequences. To determine the efficiency of barcode indexing, we determined the number of sequences of each barcode.
Results
Efficiency of barcode indexing
Using barcode indexing, the 16 barcode-indexed sets with each set containing six samples from patients with PD were mixed in one tube and subjected to sequence analysis using the SOLiD System. Using one of the eight lanes of the slide, the SOLiD System generated 9 836 344 reads (50 bp in length), 9 712 211 reads (98.7%) of which carried the sequences of one of the 16 barcode-tagged sequences. Subsequently, 5 992 551 reads (59.2%) of the barcode-tagged reads were aligned to the reference sequences under the conditions that allow up to two color space mismatches (Figure 2). Comparable average numbers of aligned reads for individual bases from 1513 to 3917 were obtained among the barcode sets, which are sufficient for the identification of variants in the pooled DNAs from six individuals.

Data of 16 barcode-indexed sets. Total number of reads and those of individual barcode-indexed reads are shown.
Efficiency of detection of variants in pooled DNAs
To detect the rarest single-nucleotide variants (SNVs) of 1 in 12 alleles whose fraction is expected to be 0.083, we attempted to determine the optimal settings for the threshold of the fraction of variant reads. In the sequenced regions of GBA in the 96 patients with PD, there were 99 nucleotides containing SNVs in total, as confirmed by Sanger sequencing with various frequencies from 1 to 12 variant alleles among the 12 alleles in the 16 sets of pooled DNAs (Table 1). Sufficient read depths of 1912–6732 were obtained for the 99 nucleotides containing bona fide SNVs. For the 99 nucleotides containing bona fide SNVs, the observed and predicted frequencies of variant alleles highly correlated (correlation coefficient r2=0.994) across the wide range of frequencies: from 1 to 12 alleles among the 12 alleles (Figure 3). However, the error rate for individual nucleotides without SNVs considerably varied, and 503 of the 68 149 nucleotides (0.74%) showed exceptionally high error rates exceeding 0.02, despite a low average error rate (0.00235). In addition, the fraction of variant nucleotides in the 27 nucleotides containing a single-variant allele among the 12 alleles (predicted frequency of 0.083) also varied (0.024–0.089) to a considerable degree, making it difficult to distinguish a nucleotide with a single bona fide variant from the nucleotides without SNVs with high error rates exceeding 0.02. To detect the rarest SNVs (1 among 12 alleles) without losing any of these SNVs, we considered that the threshold should be set at 0.02. When the threshold to detect variants was set at 0.02, as many as 503 false-positive SNVs were simultaneously detected. As the number of false-positive SNVs is too large, an efficient filtering system to eliminate as many as false-positive SNVs should be established. To accomplish this, the properties of the sequence reads of the false-positive SNVs were investigated. In this study, we specifically focused on cycle number-dependent errors (position in the sequencing read) and sequence context-dependent errors.

The number of variant alleles among 12 alleles and the observed fraction of the number of variant reads. The horizontal axis represents the number of variant alleles among 12 alleles and the vertical axis represents the observed fraction of the number of variant reads. The scatter plot of each nucleotide is dotted in the bona fide variants (number of variant alleles of 1–12).
Cycle number-dependent errors
First, nucleotides whose error rates exceeded 0.01 (1784 nucleotides) were analyzed to obtain the fraction of the number of variant reads to the total number of reads at each cycle, and it was found that the cycles of 11–35 bases of each read contained fewer errors than the first 10 bases or the last 15 bases (Figure 4a). In contrast, the fractions of the number of variant reads were independent of the sequence cycles in the bona fide SNVs (Figure 4b). Thus, bases at 11–35 cycles were used to identify SNVs, reducing the average read depths from 2746- to 1626-fold, which seemed to be still sufficient for further analysis. Under this condition, all the bona fide SNVs (99 nucleotides) were still detected without losing any of these SNVs and the number of false-positive SNVs was reduced from 503 to 173 (Tables 2 and 3).

Ratios of number of variant reads to total number of reads at each sequence cycle. Ratios of the number of variant reads to the total number of reads at each sequence cycle in nucleotides whose error rates exceeded 0.01 (a) and bona fide variants (b).
Sequence context-dependent errors
The sequence context-dependent errors are considered to be reflected as the directional bias in the reads including ‘variant nucleotides,’ and we defined ‘strand bias’ as the ratio of the number of forward sequence reads to the total number of sequence reads including variant nucleotides. At each nucleotide, the reads containing ‘variant nucleotides’ at bases 11–35 were collected and strand bias was calculated. It was revealed that the strand biases in the false-positive SNVs (error rate exceeding 0.02) were much higher than those in the bona fide variants (Figure 5), whereas the strand biases in the bona fide variants ranged from 0.17 to 0.76. Thus, when limiting the strand bias between 0.17 and 0.76 to identify SNVs, the number of false-positive SNVs was reduced from 173 to 27, whereas all the bona fide variants (99 nucleotides) were still detected (Tables 2 and 3).

Strand bias. Ratios of the number of forward sequence reads to the total number of sequence reads including variant nucleotides were calculated in the false-positive SNVs (error rate exceeding 0.02) and the bona fide variants.
Discussion
Genome-wide association studies based on the common disease-common variants hypothesis have been widely used to detect disease-susceptibility genes, but the odds ratios of the genes identified by this approach are generally low. In contrast, the genes identified on the basis of the ‘common disease-multiple rare variants’ are considered to have large effect sizes.3, 4, 5, 6 To identify all the rare variants, comprehensive resequence analyses of candidate genes or eventually all the genes in the human genome are required. To accomplish this aim, efficient methods of resequencing genes in a large number of samples should be established. There are several methods of multiplexing samples to be analyzed, such as pooling of samples, barcode indexing and physical separation (segmentation) of samples on a slide. By combining these protocols, hundreds or thousands of samples may be analyzed in a single run. In this study, using sixfold pooled samples and barcode indexing (16 plex) as well as appropriate filtering pipelines to remove false-positive SNVs, we were able to successfully detect all the 99 nucleotides containing SNVs in 1 among the 96 samples with acceptable rates of false-positive SNVs (27 nucleotides in total), confirming the usefulness of the barcode-based multiplexing using pooled DNAs for target resequencing of a large number of samples.
In this study, we found that the error rate for individual nucleotides varied and a substantial number of false-positive SNVs appeared when the threshold of the fraction of the number of variant nucleotides was set at 0.02 to detect all the bona fide SNVs of 1 among 12 alleles. Considering the aspects of constitutional error tendencies, both the cycle number-dependent errors and strand bias, which reflects sequence context-dependent errors, were analyzed. By limiting the cycle numbers to 11–35 and the strand bias to 0.17–0.76, the false-positive SNVs were markedly decreased by 95% (from 503 to 27). The parameters for error filtering should be optimized depending on the size of target sequences, read depth, multiplexity of samples and sequencer platforms. Improvement of the algorithm for sequence context-dependent errors is expected to further decrease error rate.11
Regarding pooling size, it should be taken into consideration that nucleotides with exceptionally high error rates (larger than 0.02) occur independently at a certain probability and that the fraction of the number of variant nucleotides in the rarest SNVs (predicted frequency of 0.083) also considerably varies (observed frequency of 0.024–0.089). As a consequence, increments in multiplicity of pooling by more than six will inevitably increase the rate of false-positive SNVs. Although the reasons for the underestimations of the frequency of reads containing bona fide SNVs in this study were uncertain, the possible explanations of which include technical errors of DNA quantification and/or pooling samples, the fluctuations in the relative amount of SNV alleles in the PCR steps and the stringent alignment conditions we used. For barcode indexing, it was shown that the interbarcode index difference in the number of aligned reads was relatively small. Barcode indexing has the obvious benefits of enabling the multiplexing of samples within a single run and the easy identification of individual pools with variants based on the barcode. However, library preparation of each sample set for barcode indexing remains laborious and may be a bottleneck of throughput. Although we should further optimize the protocols and algorithms for error filtering, the approach we described here is immediately applicable to the systematic exploration of multiple rare variants of candidate susceptible genes in case–control association studies and to the efficient screening for pathogenic variants of causative genes.
References
Altshuler, D., Daly, M. J. & Lander, E. S. Genetic mapping in human disease. Science 322, 881–888 (2008).
Frazer, K. A., Murray, S. S., Schork, N. J. & Topol, E. J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251 (2009).
Aharon-Peretz, J., Rosenbaum, H. & Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N. Engl. J. Med. 351, 1972–1977 (2004).
Lwin, A., Orvisky, E., Goker-Alpan, O., LaMarca, M. E. & Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 81, 70–73 (2004).
Mitsui, J., Mizuta, I., Toyoda, A., Ashida, R., Takahashi, Y., Goto, J. et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch. Neurol. 66, 571–576 (2009).
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, J., Annesi, G., Barbosa, E. R. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N. Engl. J. Med. 361, 1651–1661 (2009).
Shendure, J. & Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145 (2008).
Craig, D. W., Pearson, J. V., Szelinger, S., Sekar, A., Redman, M., Corneveaux, J. J. et al. Identification of genetic variants using bar-coded multiplexed sequencing. Nat. Methods 5, 887–893 (2008).
Koprivica, V., Stone, D. L., Park, J. K., Callahan, M., Frisch, A., Cohen, I. J. et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 66, 1777–1786 (2000).
Li, H., Ruan, J. & Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18, 1851–1858 (2008).
Druley, T. E., Vallania, F. L., Wegner, D. J., Varley, K. E., Knowles, O. L., Bonds, J. A. et al. Quantification of rare allelic variants from pooled genomic DNA. Nat. Methods 6, 263–265 (2009).
Acknowledgements
This work was supported in part by KAKENHI (Grant-in-Aid for Scientific Research) on Priority Areas, Applied Genomics, Global Center for Education and Research for Chemical Biology of the Diseases, and Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and a Grant-in-Aid for ‘the Research Committee for Ataxic Diseases’ of the Research on Measures for Intractable Diseases from the Ministry of Health, Welfare and Labour, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mitsui, J., Fukuda, Y., Azuma, K. et al. Multiplexed resequencing analysis to identify rare variants in pooled DNA with barcode indexing using next-generation sequencer. J Hum Genet 55, 448–455 (2010). https://doi.org/10.1038/jhg.2010.46
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.46
Keywords
This article is cited by
-
A labor- and cost-effective non-optical semiconductor (Ion Torrent) next-generation sequencing of the SLC12A3 and CLCNKA/B genes in Gitelman’s syndrome patients
Journal of Human Genetics (2014)
-
Evaluating rare variants under two-stage design
Journal of Human Genetics (2012)
