
Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is characterised by progressive fibro-fatty replacement of right ventricular myocardium. Earlier studies described ARVC as non-inflammatory, non-coronary disorder associated with arrhythmias, heart failure and sudden death due to functional exclusion of the right ventricle. Molecular genetic studies have identified nine different loci associated with ARVC; accordingly each locus is implicated for each type of ARVC (ARVC1–ARVC9). So far five genes have been identified as containing pathogenic mutations for ARVC. Though mutations in each of the gene/s indicate disruption of different pathways leading to the condition, the exact pathogenesis of the condition is still obscure. This review tries to understand the pathogenesis of the condition by examining the individual proteins implicated and relate them to the pathways that could play a role in the aetiology of the condition. Cardiac ryanodine receptor (RYR-2), which regulates intra-cellular Ca2+ concentration by releasing Ca2+ reserves from the sarcoplasmic reticulum (SR), was the first gene for ARVC. The mutation in this gene is believed to disrupt coupled gating of RYR-2, causing after-depolarisation, leading to arrhythmias followed by structural changes due to altered intra-cellular Ca2+ levels. Three other genes implicated for ARVC, plakoglobin (Naxos disease), desmoplakin (ARVC8) and plakophilin (ARVC9) have prompted the speculation that ARVC is primarily a disease of desmosomes. But identification of TGFβ-3 for ARVC1 and the role of all these three genes (plakoglobin, desmoplakin and plakophilin) in cardiac morphogenesis indicate some kind of signal-transducing pathway disruption in the condition. The finding that ARVC as a milder form of Uhl’s anomaly indicates similar ontogeny for the condition. Further, discovery of apoptotic cells in the autopsy of the right ventricular myocardium of ARVC patients does indicate a common pathway for different types of ARVCs, which is more specific for the right ventricular myocardium involving desmosomal plaque proteins, growth factors and Ca2+ receptors.
Similar content being viewed by others


Introduction
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is a genetically heterogenous disorder characterised by progressive fibro-fatty replacement of the right ventricular myocardium. The predominant presenting symptoms are due to ventricular arrhythmias, including palpitations, sustained ventricular tachycardia or uncommonly sudden cardiac death. Although ARVC is an unusual disorder, it has been described as the most common cause of sudden death in young athletes under the age of 35 years (Corrado et al. 1990). Heart failure is infrequent but may occur due to severe right or bi-ventricular enlargement.
Ever since the first description of the disease by Fontaine et al. (1977) as the non-inflammatory, non-coronary disorder associated with arrhythmias, heart failure and sudden death due to functional exclusion of the right ventricle among young people, extensive clinical and molecular genetic studies have been carried out. But the progress in clear understanding of the disorder has been impeded by the difficulty in diagnosis.
Diagnosis
Standardised major and minor criteria encompassing structural, histological, electrocardiographic, arrhythmic and genetic factors have been proposed in order to establish diagnosis (Table 1) (McKenna et al. 1994). According to this classification, the presence of two major criteria or one major and two minor criteria or four minor criteria are required in order to confirm ARVC.

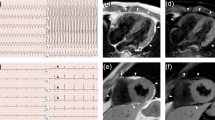
Electrocardiogram of an arrhythmogenic right ventricular cardiomyopathy (ARVC) patient showing right pre-cordial T–wave inversions
Clinical pathology
Four clinicopathologic phases have been described based on the long-term follow-up of clinical data.
Concealed phase
This phase is characterised by subtle right ventricular structural changes with or without ventricular arrhythmias during which sudden death may occasionally be the first manifestation of the disease, mostly in young people during competitive sports or intense physical exercise (Thiene et al. 1988).
Overt arrhythmic phase
This phase shows symptomatic right ventricular arrhythmias possibly leading to sudden cardiac arrest associated with overt right ventricular functional and structural abnormalities (Corrado et al. 1990).
Global right ventricular dysfunctional phase
This phase results from the progression and extension of muscle disease with relatively preserved left ventricular function (Corrado et al. 1998).
Final phase
The final stage of bi-ventricular pump failure is caused by pronounced left ventricular involvement. At this stage, ARVC mimics biventricular dilated cardiomyopathy of other causes, leading to congestive heart failure and related complications.
Molecular genetics
Various pathological and familial studies have described nine types of ARVC, the types being classified based on different loci mapped for the condition (Table 2). Nine different loci for the autosomal dominant form of ARVC indicate the underlying genetic heterogeneity. Among them, five genes have been identified as containing the causative mutations for ARVC. Other genes implicated in ARVC have to be identified in order to elucidate the pathogenic pathway leading to the condition.
Genes identified
Cardiac ryanodine receptor (RYR-2)
The first gene identified is RYR-2, the cardiac ryanodine receptor, mutations in which cause the characteristic effort-induced arrhythmias associated with ARVC-2 (Nava et al. 1988). The critical region was identified by the mapping studies carried out by Rampazzo et al. (1995) and Bauce et al. (2000). Physical mapping of the critical region of ARVC2 elucidated the pathogenic role of mutations in the RYR-2 gene (Tiso et al. 2001).
The most important regulatory pathway that is believed to be disrupted in ARVC is mediated by FKBP 12.6. The binding of FKBP 12.6 mainly causes coupled gating among the Ca2+ release channels, which allows simultaneous opening and closing of all RYR2 channels on the membrane (Marx et al. 1998). FKBP thereby controls the opening of RYR2 in response to any trigger. This process is very important for regulating the cytosolic [Ca2+], required for terminating the muscle contraction by reducing [Ca2+] required for relaxation and preventing diastolic depolarisation, which can cause ventricular arrhythmias. Therefore, FKBP 12.6 binding makes RYR2 refractory to any trigger for Ca2+ release.
Two of the four mutations detected in the ARVC2 patients occur in FKBP 12.6 binding domain (Tiso et al. 2001). This may make the binding of FKBP12.6 less stable, resulting in loss of coupled gating and causing after-depolarisation, leading to arrhythmias. Also, the unstable nature of RYR-2 resulting from lack of proper binding of FKBP 12.6 may cause massive leakage of Ca2+ from RYR2, even on minor perturbations such as stress (Tiso et al. 2001).Thus, the arrhythmias found in the ARVC patients could be explained. Structural changes accompanying the arrhythmias can be explained by the reports that imbalance of intra-cellular Ca2+ levels can trigger apoptosis, which is identified as a primary mechanism for cardiac muscle degeneration in ARVC (Mallat et al. 1996; Valente et al. 1998).
The four other genes identified in ARVCs are desmoplakin (ARVC8), plakophilin (ARVC9), plakoglobin (Naxos) and TGFβ-3 (ARVC1). All the genes except TGFβ-3 are involved in formation of desmosomal plaque, leading to the suggestion that ARVC is primarily a disease of desmosomes (Fig. 2) (Gerull et al. 2004).

Schematic representation of desmosomal structure
Desmoplakin
The second gene identified for ARVCs was desmoplakin, associated with ARVC8. Rampazzo et al. (2002) identified S299R in exon 7 of DSP as the pathogenic mutation in a family of 26 members spanning four generations. Desmoplakin is exclusive to desmosomes and is a ubiquitous member of all the desmosomal complexes found in various tissues. It is the key protein of inner-most desmosomal plaque, serving as an attachment site for cytoplasmic intermediate filaments apart from playing a role in desmosomal assembly, stability and regulation and affecting the modeling of tissue architecture during embryogenesis (Gallicano et al. 1998). Mutations in desmoplakin have been reported for various disorders of skin and heart, which are striate palmoplantar keratoderma II (SPPK2) (Armstrong et al. 1999), dilated cardiomyopathy with woolly hair and keratoderma (DCWHK) (Norgett et al. 2000), skin fragility–woolly hair syndrome (SFWHS) (Whittock et al. 2002) and familial ARVC8.
It can be concluded from the causative mutations for these disorders that absence of the segment of the tail region at the C terminal causes dilated cardiomyopathy. Therefore, the mutations causing the improper interactions with intermediate filaments or plakoglobin have been tolerated by the heart muscle, as seen in skin disorders of DSP (SPPK2, SFWHS), and only complete absence of the segment or presence in only one copy would lead to heart abnormalities.
In ARVC8, there is a ser299-to-arg (S299R) mis-sense mutation in exon 7 of the DSP gene. This mutation is located in the N-terminal region modifying the putative PKC phosphorylation site in the minor heptad-rich Z region between the two alpha helical segments forming a loop that interacts with plakoglobin. Unlike all the other desmoplakin related disorders, there is no skin abnormality associated with the mutation in ARVC. The epidermal tissue and the left ventricular myocardium seem to have tolerated the mutation, as only the right ventricle is affected. This particular mutation seems to play a very important role in the right ventricular myocardium as it is inherited as autosomal dominant disorder. The predilection for the right ventricle is yet to be understood.
There seems to be functional redundancy for C-terminal domain with some other proteins, as in Carvajal syndrome, single dosage seems to be enough to show normal phenotype. But manifestation of the N-terminal S299R in ARVC shows that no functional redundancy of the N-terminal globular domain is available in the right ventricular myocardial tissue. Absence of keratoderma phenotype in ARVC can be explained by considering that only the mutations in C-terminal regions affect the skin phenotype, as seen in various disorders.
Plakoglobin (JUP) (DSPIII) (γ-catenin)
Initially, plakoglobin was identified as the gene involved in the aetiology of ARVC through mapping studies for Naxos disease, which localised the gene to 17q21. Naxos disease constitutes the triad of ARVC, diffuse, non-epidermolytic palmoplantar keratoderma and woolly hair and was originally described by Protonotarios et al. (1986) in the Greek island of Naxos. Studies on nine families with 21 affected individuals by Coonar et al. (1998) initially mapped the gene to 17q21. McKoy et al. (2000) identified homozygous 2-bp deletion in the plakoglobin gene in 19 affected individuals. Plakoglobin is found in desmosomes, adherens junction and cytosol and is involved in regulating junction assembly and implementing morphoregulatory signals in the Wnt pathway. It is a major desmosomal protein, and its role in the adherens junction has been recently reported (Sacco et al. 1995).
Only one gene with armadillo repeat has been found in Drosophila, and evolution must have caused the divergence of this gene into beta-catenin and plakoglobin (White et al. 1998). So, the functions performed by these two genes in the mammalian system are performed by a single gene in Drosophila. This gives an indication that one protein can perform the functions of the other in its absence due to functional redundancy, thereby alleviating the effect of the absence of one of them. This may explain the requirement of the mutant gene in double dose for ARVC in Naxos as opposed to single dose requirement in ARVC8. Further, it has been reported that despite their normal participation in different junctions, both these proteins can suffice the adhesion function of armadillo in Drosophila, and in gamma catenin (plakoglobin) null mouse embryos, it has been found that beta-catenin localises to desmosomes (Ruiz et al. 1996; Bierkamp et al. 1999).
It has been established that though central armadillo repeats interact with various proteins, it is the COOH domain of plakoglobin that is involved in transcription activation (White et al. 1998). The 2-bp deletion identified in Naxos eliminates nucleotides 2,157 and 2,158, causing shift in the reading frame and resulting in the premature termination and loss of 56 amino acids in the COOH– terminal end. Since it is the –COOH end that is implicated in the signalling function, the pathogenesis of ARVC may include some of the signalling activities disturbed along with the myocyte integrity.
If the signalling function of these proteins is considered to be involved in the pathogenesis, along with growth factors which may have a role in the Wnt pathway, all the other desmosomal plaque proteins with signal-transducing activity could be considered as candidate genes for ARVC. Immunochemical studies have found band 6 (a protein closely related in structure to plakoglobin) desmocalmin and desmoyokin as the minor components whose interaction with the cytoplasmic domain of desmosomal cadherins and roles in various signalling function have not been completely defined (Tsukita and Tsukita 1985; Kapprell et al. 1988).
Plakophilin2 (pkp2)
Plakophilin has been recently identified as the third desmosomal plaque protein involved in the pathogenesis of ARVC, and the disorder was named as ARVC9 by Gerull et al. (2004). Like armadillo of Drosophila, plakophilins also have a role in embryonic development, as shown by studies on pkp2 null mice mutants by Grossmann et al. (2004). The mice show lethal alterations in the morphogenesis and architecture of the heart with reduced trabeculation, disarrayed cytoskeleton and rupture of cardiac walls. Apart from developmental abnormalities, absence of pkp2 also affects the desmosomal assembly. In the cardiac tissue devoid of pkp2, desmoplakin dissociates from the desmosomal plaque and forms granular aggregates in the cytoplasm, thereby affecting the connection of cardiomyocytes. It is speculated that cell adhesion function is disrupted in cardiomyocytes as a result of mutations in plakophilin2, but the involvement of the signalling function of plakophilins in the pathogenesis cannot be ruled out.
TGFβ-3
Transforming growth factors ß1, 2, 3 (TGF-ß1, 2, 3) are generally described as the pleiotropic cytokines, with regulatory roles in the process of tissue repair and remodelling following injury. Beffagna et al. (2005) detected regulatory mutations in the TGFβ-3 gene in 5′UTR region resulting in a twofold increase in expression at 14q24.3, a cytogenetic locus for ARVC1. Among various functions, TGFβ-3 is involved in the regulation of angiogenesis and trophoblast differentiation during hypoxia and plays a major role in cardiac morphogenesis.
Studies on all the three TGFs were carried out on knockout mice in order to determine their roles in morphogenesis of the heart (Molin et al. 2003). TGFβ-3 has roles in mesenchyme differentiation, development of fibrous septum primum of the atrium and fibrous skeleton of the heart. The major role of TGFβ-3 in differentiation of the fibrous structures of the heart has recently been elucidated. The twofold increase in TGFβ-3 synthesis may play a role in excess fibrosis (along with fatty replacement) observed in ARVC, thus providing the basis for the trans-differentiation theory (discussed below). A foetal gene program must have been revoked in the adult heart, leading to the replacement of the normal myocardium, resulting in abnormal fibrous skeleton of the heart. Arrhythmias observed in the case of ARVC could have been the result of these perturbations, as the cardiac fibrous skeleton mainly serves in maintaining the electrophysiological discontinuity between the atria and ventricles (only connection via the atrioventricular bundle is allowed).
Molecular pathogenesis
Though the genes and causative mutations have been identified in the case of ARVC8 (Desmoplakin), Naxos (Plakoglobin), ARVC2 (RYR-2), ARVC9 (plakophilin) and ARVC1 (TGFβ-3), the pathogenesis of the condition is obscure. The genes with diverse functions, such as desmosomal plaque proteins (involved in cell adhesion) and ryanodine receptor (regulates intra-cellular Ca2+ concentration) and TGF (involved in morphogenesis and tissue remodelling) are implicated in causing different types of ARVC with similar histopathology and electrophysiology. This indicates that a common pathway operates involving these proteins in early predilection to the right ventricle though all these genes except RYR-2 are expressed in many other tissues. Also, RYR-2 is not specific to the right ventricle alone, but the pathway seems to involve the FKBP binding domain in conjunction with all the other proteins in the pathogenesis.
The implication of calcium channels and growth factors in the disease suggests that the mechanism may not be as simple as disruption of tissue integrity due to improper desmosomal plaque formation by the mutated proteins, resulting in myocyte loss. The mechanism of myocyte loss may be due to complex interactions specific to the right ventricle, with many growth factors and intra-cellular ionic concentration playing a role.
Various theories have been proposed for explaining the aetiology of the condition which include:
-
(1)
The dysontogenetic theory
-
(2)
The trans-differentiation theory
-
(3)
The apoptotic theory.
The dysontogenetic theory (Ananthasubramaniam and Khaja 1998) suggests that ARVC is a milder form of “parchment RV” or Uhl’s anomaly, a congenital hypoplasia of the RV myocardium, which presents in infancy as congestive heart failure (CHF). Uhl’s anomaly is characterised by partial or complete absence of the myocardium of the right ventricle. The pathogenesis of the condition has not yet been elucidated but is believed to be due to incessant apoptosis, progressively causing complete loss of the myocardium.
The pathogenic mechanism may involve the natural process that causes disparity between the thickness of left and right ventricles (James et al. 1996). There is a normal reduction in pressure against which the right ventricle must pump as soon as the heart of the new born starts working. So an adaptive mechanism operates that causes differential thickness of both ventricles. Two alternate mechanisms have been suggested: One of them states that thickening of the left ventricle occurs in order to compensate the increased haemodynamic burden and right ventricle remains stable; the other one explains the cause of discrepancy in thickness as being due to the removal of surplus tissue and cells by the body. The second mechanism seems to operate in this post-natal right ventricular morphogenesis since a similar process occurs in the post-natal morphogenesis of the brain, where an excess of neurons are removed by apoptosis. De-regulation of this process, which limits apoptosis to the extent that the haemodynamic pressure is maintained, may lead to complete loss of the myocardium (James et al. 1996). This may be the mechanism that operates in the case of Uhl’s anomaly, where there is failure of cessation of apoptotic signals or anti-apoptotic signals fail to operate in order to rescue the right ventricular myocardium from complete loss.
The pathogenesis of ARVC also could involve a similar mechanism, but the difference could be that in Uhl’s anomaly, this process could end in complete destruction of the right ventricular myocardium, and in ARVC, this process would be episodic focal apoptosis beginning at any time in life and may re-occur, which may lead to sudden death. The arrhythmias that appear in ARVC may be due to the cells undergoing apoptosis, which may transiently exhibit increased excitability, or the normal route of electrical signal transmission may be altered due to the apoptotic cells leading to micro-re-entrant or macro-entrant circuits.
This hypothesis of apoptosis has been supported by the recently propounded apoptotic theory. Apoptosis in ARVC has been shown by Mallat et al. (1996) from the autopsy of the right ventricular myocardium. An interesting observation was that the cells undergoing apoptosis were most frequently found in areas that have not undergone fibro-fatty replacement, and very rarely, the apoptotic cells were observed in the areas already affected. This shows that apoptosis could be the primary process that precedes fibro-fatty replacement of myocardial tissue. Further elucidation of apoptosis as the primary process would require determining the triggering factors for apoptotic myocardial cell death. If the theory of apoptotic post-natal morphogenesis is to be considered as involved in pathogenesis, then the factors involved in this pathway are to be elucidated, and also, the question as to why apoptotic loss of the myocardium occurs intermittently in ARVC as opposed to progressive and complete loss in Uhl’s anomaly needs to be addressed.
A proposed mechanism alternative to or in addition to apoptosis is transdifferentiation. The transdifferentiation theory is based on the hypothesis that myocardial cells can change from muscle to fibrous and adipose tissue and the observation that in one patient, “transitional cells” at the interface between cardiac muscle and adipose tissue expressed both desmin, which is characteristic of muscle tissue, and vimentin, expressed only in adipocytes (D’amati et al. 2000).
Another study by Yamamoto et al. (2000) showed a different pattern of apoptosis in the biopsies of right ventricular myocardium from eight patients with ARVC. Along with the normal nuclear apoptosis, developmental programmed cell death was observed, which includes nuclear migration and extrusion. This supports the hypothesis that a developmental gene program is revoked in this condition. This gene program, which would operate just after birth (for eliminating excess of right ventricular tissue) seems to have been revoked in ARVC at different intervals during the lifetime of the affected individual. Undoubtedly, the trigger for ARVC is different from that of Uhl’s anomaly, and the effect of this factor seems to be episodic.
With the background of these theories, a pathway needs to be elucidated that should include desmosomal plaque proteins such as plakoglobin, desmoplakin and plakophilin, calcium channel and the cardiac ryanodine receptor, and growth factors such as TGFβ-3.
In all the cases where the genes have been identified, pathogenesis can be partially explained. For instance, in the case of ARVC2, RYR-2 deregulation due to mutations in the FKBP 12.6 binding domain could explain effort-induced arrhythmias. Also, the role of calcium signalling in apoptosis could be implicated in myocyte loss, but the predilection of this loss to the right ventricle could not be explained. Mutations in the C-terminal end of RYR-2 are known to cause catecholaminergic polymorphic ventricular tachycardia (CPVT), which includes arrhythmias without structural changes. If the myocyte loss was only due to cytoplasmic calcium overload, myocyte degeneration would have accompanied CPVT. So some other pathway involving only the amino terminal domain would result in myocyte loss.
Mutation in the case of ARVC8 (S299R) in exon 7 of desmoplakin-modifying putative phosphorylation by PKC disrupts its interaction with plakoglobin. This interaction may have another role in addition to the desmosomal assembly, involving some kind of signalling pathway.
Plakoglobin and plakophilin have been directly demonstrated to have a role in morphogenesis of the heart. Since both are armadillo-like proteins, it has to be further studied whether the Wnt pathway has any role in post-natal right ventricular morphogenesis.
The pathway involved in post-natal morphogenesis of the right ventricle should have desmosomes or desmosomal plaque proteins as its components, with a trigger from growth factors such as TGFβ-3 and dependence on intra-cellular calcium concentration through interactions with amino terminal domain of the RYR-2 receptor. Understanding the pathogenesis of this process would have instructional value in understanding additional mechanisms of developmental regulation of the heart, apart from providing therapeutic significance.
References
Ananthasubramaniam K, Khaja F (1998) Arrhythmogenic right ventricular dysplasia/cardiomyopathy: review for the clinician. Prog Cardiovasc Dis 41:237–246
Armstrong DK, McKenna KE, Purkis PE, Green KJ, Eady RAJ, Leigh IM, Hughes AE (1999) Haploinsufficiency of desmoplakin causes a striate subtype of palmoplantar keratoderma. Hum Mol Genet 8:143–148
Bauce B, Nava A, Rampazzo A, Daliento L, Muriago M, Basso C, Thiene G, Danieli GA (2000) Familial effort polymorphic ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy map to chromosome 1q42-q43. Am j Cardiol 85:573–579
Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A (2005) Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res 65(2):366–373
Bierkamp C, Schwarz H, Huber O, Kemler R (1999) Desmosomal localization of -catenin in the skin of plakoglobin null-mutant mice. Development 126:371–381
Coonar AS, Protonotarios N, Tsatsopoulou A, Needham EW, Houlston RS, Cliff S, Otter MI, Murday VA, Mattu RK, McKenna WJ (1998) Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 97(20):2049–2058
Corrado D, Thiene G, Nava A, Rossi L, Pennelli N (1990) Sudden death in young competitive athletes: clinicopathologic correlation in 22 cases. Am J Med 89:588–596
Corrado D, Basso C, Schiavon M, Thiene G (1998) Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 339:364–369
D’amati G, Di Gioia C, Giordano C, Gallo P (2000) Myocyte transdifferentiation: a possible pathogenetic mechanism for arrhythmogenic right ventricular cardiomyopathy. Arch Pathol Lab Med 124:287–290
Fontaine G, Frank R, Vedel J, Grosgogeat Y, Cabrol C, Facquet J (1977) Stimulation studies and epicardial mapping in ventricular tachycardia: study of mechanisms and selection for surgery. In: Kulbertus HE (eds) Reentrant Arrhythmias. MTP Publishing, Lancaster, pp 334–350
Gallicano GI, Kouklis P, Bauer C, Yin M, Vasioukhin V, Degenstein L, Fuchs E (1998) Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol 143:2009–2022
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, Peters S, Grossmann KS, Michely B, Sasse-Klaassen S, Birchmeier W, Dietz R, Breithardt G, Schulze-Bahr E, Thierfelder L (2004) Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 36:1162–1164
Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW, Birchmeier W (2004) Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol 167:149–160
James TN, Nichols MM, Sapire DW, DiPatre PL, Lopez SM (1996) Complete heart block and fatal right ventricular failure in an infant. Circulation 93:1588–600
Kapprell HP, Owaribe K, Franke WW (1988) Identification of a basic protein of Mr 75,000 as an accessory desmosomal plaque protein in stratified and complex epithelia. J Cell Biol 106:1679–1691
Mallat Z, Tedjin A, Fontaliran F, Fontaine G (1996) Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med 335:1190–1196
Marx SO, Ondrias K, Marks AR (1998) Coupled gating between individual skeletal muscle Ca21 release channels (ryanodine receptors). Science 281:818–821
McKenna WJ, Thiene G, Nava A et al (1994) Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J 71:215–218
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, Norman M, Baboonian C, Jeffery S, McKenna WJ (2000) Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355:2119–2124
Molin DG, Bartram U, Van der Heiden K, Van Iperen L, Speer CP, Hierck BP, Poelmann RE, Gittenberger-de-Groot AC (2003) Expression patterns of Tgfbeta1–3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Dev Dyn 227(3):431–444
Nava A, Canciani B, Daliento L, Miraglia G, Buja G, Fasoli G, Martini B, Scognamiglio R, Thiene G (1988) Juvenile sudden death and effort induced ventricular tachycardias in a family with right ventricular cardiomyopathy. Int J Cardiol 21:111–123
Norgett EE, Hatsell SJ, Carvajal-Huerta L, Cabezas J-CR, Common J, Purkis PE, Whittock NV, Leigh IM, Stevens HP, Kelsell DP (2000) Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 9:2761–2766
Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S, Scampardonis G (1986) Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J 56:321–326
Rampazzo A, Nava A, Erne P, Eberhard M, Vian E, Slomp P, Tiso N, Thiene G, Danieli GA (1995) A new locus for arrhythmogenic right ventricular cardiomyopathy(ARVD2)maps to chromosome 1q42-q43. Hum Mol Genet 4:2151–2154
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, Zimbello R, Simionati B, Basso C, Thiene G, Towbin JA, Danieli GA (2002) Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 71:1200–1206
Ruiz P, Brinkmann V, Ledermann B, Behrend M, Grund C, Thalhammer C, Vogel F, Birchmeier C, Gunthert U, Franke WW (1996) Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol 135:215–225
Sacco PA, McGranahan TM, Wheelock MJ, Johnson KR (1995) Identification of plakoglobin domains required for association with N-cadherin and a-catenin. J Biol Chem 270:20201–20206
Thiene G, Nava A, Corrado D, Rossi L, Pennelli N (1988) Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 318:129–33
Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A (2001) Identification of mutations in cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2(ARVD 2). Human Mol Genet 10(3):189–194
Tsukita S, Tsukita S (1985) Desmocalmin: a calmodulin-binding high molecular weight protein isolated from desmosomes. J Cell Bid 101:2070–2080
Valente M, Calabrese F, Thiene G, Angelini A, Basso C, Nava A, Rossi L (1998) Invivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol 152:479–484
White P, Aberle H, Vincent JP (1998) Signaling and adhesion activities of mammalian b-catenin and plakoglobin in Drosophila. J Cell Biol 140:183–195
Whittock NV, Wan H, Morley SM, Garzon MC, Kristal L, Hyde P, McLean WHI, Pulkkinen L, Uitto J, Christiano AM, Eady RAJ, McGrath JA (2002) Compound heterozygosity for non-sense and mis-sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol 118:232–238
Yamamoto S, Tsyplenkova VG, James TN (2000) Morphological patterns of death by myocytes in arrhythmogenic right ventricular dysplasia. Am J Med Sci 320(5):310–319
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dokuparti, M.V.N., Pamuru, P.R., Thakkar, B. et al. Etiopathogenesis of arrhythmogenic right ventricular cardiomyopathy. J Hum Genet 50, 375–381 (2005). https://doi.org/10.1007/s10038-005-0273-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-005-0273-5
