
Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a fatal genetic heart disease characterized by cardiac arrhythmias, in which fibrofatty deposition leads to heart failure, with no effective treatments. Plakophilin 2 (PKP2) is the most frequently mutated gene in ARVC, and although altered RNA splicing has been implicated, there are no models to study its effect and therapeutics. Here, we generate a mouse model harboring a PKP2 mutation (IVS10-1G>C) affecting RNA splicing, recapitulating ARVC features and sudden death starting at 4 weeks. Administering AAV-PKP2 gene therapy (adeno-associated viral therapy to drive cardiac expression of PKP2) to neonatal mice restored PKP2 protein levels, completely preventing cardiac desmosomal and pathological deficits associated with ARVC, ensuring 100% survival of mice up to 6 months. Late-stage AAV-PKP2 administration rescued desmosomal protein deficits and reduced pathological deficits including improved cardiac function in adult mice, resulting in 100% survival up to 4 months. We suggest that AAV-PKP2 gene therapy holds promise for circumventing ARVC associated with PKP2 mutations, including splice site mutations.
Similar content being viewed by others

Main
ARVC is an incurable genetic heart disease that was first characterized as affecting only the right ventricle; however, increasing evidence highlights that there are left-dominant and biventricular forms of ARVC as well1,2,3. ARVC is classically characterized by early electrical defects, with a high frequency of ventricular arrhythmias that can be exacerbated with exercise, leading to sudden cardiac death1,2. However, structural defects are equally important, as ARVC progresses from an electrical phase to a structural phase in which there is fibrofatty replacement of the myocardium, leading to ventricular dysfunction and failure1,2,3. ARVC occurs in 1 out of 2,000–5,000 people, though its prevalence may be higher owing to poor diagnostic markers1,4. To date there are no effective treatments or cures for ARVC5,6,7. Current approaches are directed towards symptomatic relief and centered around lifestyle changes (for example, avoiding competitive sports that can trigger sudden cardiac death) and pharmacological interventions that target the electrical defects (for example, anti-arrhythmics drugs and beta-blockers), but also more invasive measures (for example, implantable cardioverter defibrillators, cardiac catheter ablation and heart transplantation), if the patient becomes intolerant or unresponsive to pharmacotherapy5,7. However, implantable cardioverter defibrillators have frequent device-related or lead-related complications, catheter ablations are subject to recurrence due to the generation of arrhythmogenic foci, and heart transplantation has a 23% mortality rate 10 years post procedure7. These factors highlight the critical need to identify therapeutic strategies that target the underlying drivers of the pathogenesis of ARVC.
Human genetic studies show that 40–50% of patients with ARVC carry mutations in genes of the desmosome8, which is a cell–cell adhesion structure that acts as a mechanical anchor and is therefore critical in tissues undergoing constant mechanical stress, such as cardiac muscle tissue8,9. Five classic genes make up the desmosome: desmoplakin (DSP), desmoglein 2 (DSG2), desmocollin 2 (DSC2), plakoglobin (JUP) and plakophilin 2 (PKP2), all of which have been implicated in human ARVC7,8. At the cardiac cell–cell junction, desmosomes function alongside the fascia adherens junction (which links the cell membrane to the actin cytoskeleton) and gap junctions (involved in electrical coupling) to coordinate muscle contraction9. Therefore, instrumental to understanding the pathogenesis of ARVC in the context of human genetics is that a single desmosomal gene mutation has a devastating ‘domino’ effect on the loss of expression of adjacent desmosomal proteins and neighboring gap junction proteins7,10. Although the neighboring fascia adherens junction proteins are initially spared, there is evidence that they can be affected in late stages of heart failure10, highlighting that ARVC is a disease of the desmosome with multi-protein consequences at the cell–cell junction.
PKP2 mutations have been reported to make up the majority of desmosomal gene mutations in ARVC7,8. Mechanistically, studies of patients with ARVC suggest that a variety of PKP2 mutations (insertions or deletions, nonsense, missense) lead to haploinsufficiency and therefore a reduction in myocardial PKP2 protein levels11,12,13. An effect on PKP2 RNA has also been reported, as myocardial PKP2 transcript levels were also found to be downregulated in patients with ARVC harboring PKP2 mutations13,14. More direct evidence of RNA involvement is suggested by studies that show that altered RNA splicing may also be a critical mechanism through which PKP2 patient genetics drive ARVC11,15, highlighting that altered PKP2 RNA levels may be a critical trigger of ARVC pathogenesis in patients with ARVC who have PKP2 mutations. Defects in RNA splicing have been linked to approximately one-third of all human disease-causing mutations16, further highlighting the relevance of studying this mechanism in human ARVC. Splice acceptor sites are splicing elements located at the 3′ intron–5′ exon boundary and are essential for the excision of introns to generate a mature mRNA17. Mutations in these regions are linked to many human diseases, such as arthrogyrposis multiplex congenita, Charcot–Marie–Tooth disease and Fabry disease17,18,19,20. Splice acceptor site mutations trigger aberrantly spliced transcripts through various mechanisms, including the use of a cryptic intronic splice site, exon skipping or the use of a cryptic exonic splice site, resulting in diverse effects on protein quality and levels15,18,19,20. However, there are no models and limited mechanistic insights exist into how human mutations in RNA splicing affect PKP2 biology and ARVC, and into the therapeutics or interventions that would be most impactful.
Here, through the generation of a PKP2 genetic mouse model, we highlight the sufficiency and molecular mechanisms by which a prevalent human PKP2 RNA splice acceptor site mutation (PKP2 IVS10-1G>C) triggers the postnatal onset of ARVC. PKP2 homozygous mutant (Hom) hearts displayed altered PKP2 RNA splicing, resulting in low levels of a higher molecular weight mutant PKP2 protein in the absence of endogenous PKP2 protein levels. These molecular consequences were sufficient to trigger an early and progressive disruption of the desmosome and development of all classic ARVC features (sudden cardiac death, arrhythmias, biventricular dysfunction and fibrofatty replacement of myocardium). In vitro studies showed that interventions focused on restoring PKP2 levels, irrespective of wild-type or mutant PKP2, were sufficient to prevent desmosomal protein disruption in PKP2 Hom neonatal mouse cardiomyocytes. In vivo studies leveraging an adeno-associated viral (AAV) gene therapy strategy to drive cardiac expression of PKP2 (AAV-PKP2) at early and late disease stages in PKP2 Hom mice found that it was sufficient to restore PKP2 protein dose, scaffold the desmosome and prevent ARVC-related deficits, including prolonging lifespan. These data suggest that therapeutic approaches targeted at restoring PKP2 RNA and protein levels (via PKP2 gene therapy) may have broad applicability to circumvent ARVC deficits associated with PKP2 mutations, including splice site mutations.
Results
PKP2 splice site mutation can recapitulate ARVC in mice
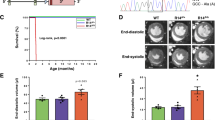
Previous studies identified a PKP2 RNA splice acceptor site mutation (PKP2 IVS10-1G>C) in multiple ARVC populations in which patients displayed classic ARVC disease features11,15,21,22, contributing to the idea that alterations in PKP2 RNA splicing may be a critical mechanism through which PKP2 patient genetics drive ARVC. This mutation is also highly relevant as it ranks at the top of the list of most frequent PKP2 mutations in ARVC cohorts in the Atlas of Cardiac Genetic Variation23. To investigate the direct consequence of the PKP2 RNA splice site IVS10-1G>C mutation in the pathogenesis of ARVC, we created a knock-in mouse model within mouse intron 9 (mouse IVS9-1G>C equivalent to human IVS10-1G>C) through CRISPR–Cas9 genome editing (Fig. 1a). Founder mice harboring the PKP2 IVS10-1G>C mutation were backcrossed three generations to remove CRISPR–Cas9-associated off-target effects and generate PKP2 IVS10-1G>C heterozygous (Het) mutant mice. All offspring were viable and born at Mendelian ratios (Supplementary Table 1); however, PKP2 IVS10-1G>C Hom mice displayed sudden death beginning at 4 weeks of age, with a median survival of 11 weeks, and no PKP2 Hom mice survived past 26 weeks of age (Fig. 1b). PKP2 Het mice also showed survival defects, although not as severe as those of PKP2 Hom mice (Fig. 1b). Telemetry electrocardiogram (ECG) recordings revealed that conscious PKP2 Het mice displayed baseline electrophysiological abnormalities (marked by the presence of premature ventricular contractions (PVCs)) in the absence of cardiomyopathy, as assessed by in vivo magnetic resonance imaging (MRI), which may account for premature lethality and sudden cardiac death at later ages (Extended Data Fig. 1). To better understand the onset of sudden cardiac death in PKP2 Hom mice (4 weeks of age), we quantitatively assessed cardiac dimensions and function in the left and right ventricles of 4-week-old PKP2 Hom mice and littermate controls using MRI. Representative four-chamber and short-axis views highlighted biventricular dilation in PKP2 Hom mice compared to littermate controls (Fig. 1c). No significant differences in heart rate were observed between mice (Fig. 1d); however, a significant decrease in left and right ventricular ejection fraction was found in PKP2 Hom mice compared to controls (Fig. 1d). Defects in cardiac function were attributed to the significantly enlarged right ventricular end-diastolic and end-systolic volumes (Fig. 1d), as well as significantly increased left ventricular end-systolic volumes in PKP2 Hom mice (Fig. 1d), highlighting biventricular dysfunction. These results are reminiscent of those in patients harboring the PKP2 IVS10-1G>C mutation, in whom both left and right ventricular dysfunction were observed21.

a, Genomic location for PKP2 IVS10-1G>C mutation equivalent in mouse (PKP2 IVS9-1G>C), single strand oligodeoxynucleotides (ssODN) with mutation template (mutation is highlighted in red). PAM, protospacer adjacent motif, labeled in green; sgRNA, single guide RNA; WT, wild type. b, Kaplan–Meier survival analysis of PKP2 Het, PKP2 Hom and littermate control (Ctrl) mice. c, Representative cardiac four-chamber and short-axis views from MRI at end-diastole. d, Quantification of left (LV) and right (RV) ventricle end-systolic volume (ESV), end-diastolic volume (EDV), ejection fraction (EF) and heart rate using cine MRI in control (black bars) and PKP2 Hom (red bars) mice (n = 4 biologically independent animals). Data are presented as mean ± s.e.m. Two-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test. Adjusted P values, ****P < 0.00001, **P < 0.01 (EDV, RV, P = 0.0026; ESV, LV, P = 0.0074), ***P < 0.001 (ESV, RV, P = 0.0001). e, Representative composite surface ECG tracings averaged from four beats in control and PKP2 Hom mice at 4 weeks of age. Scale bars, 10 ms. f, Quantification of heart rate, PR interval and QRS interval from composite surface ECG tracings (controls, n = 4; Hom, n = 5 biologically independent animals). Data are presented as mean ± s.e.m. Two-tailed unpaired t-test. *P < 0.05 (P = 0.0198). g, Representative ECG tracings from control and PKP2 Hom mice at 4 weeks of age (controls, n = 6; Hom, n = 8 biologically independent animals). h, Quantification of mice demonstrating PVCs (right, red arrows) (controls, n = 6; Hom, n = 8 biologically independent animals). Two-tailed Fisher’s exact test. *P < 0.05 (P = 0.0310).
Electrical dysfunction is a key hallmark of ARVC and a major contributor to sudden cardiac death in patients2. To assess electrical defects in PKP2 Hom mice, we performed surface ECGs in 4-week-old mice. The analysis of composite surface ECG tracings from PKP2 Hom mice and littermate controls showed no difference in heart rate or PR intervals; however, QRS complexes were significantly widened in PKP2 Hom mice (Fig. 1e,f), indicative of ventricular depolarization abnormalities, which are common in patients with ARVC1,2,3. Ventricular depolarization abnormalities can serve as a primer for ventricular re-entry that is termed PVC24. The analysis of surface ECG tracings revealed PVCs in 60% of PKP2 Hom mice at 4 weeks of age (onset of sudden death), whereas none were observed in littermate controls (Fig. 1g,h). Telemetry ECG recordings revealed that conscious PKP2 Hom mice had isolated PVCs that precipitated into ventricular tachycardia and fibrillation (Extended Data Fig. 2), further suggesting that ventricular depolarization abnormalities can serve as a primer for life-threatening arrhythmias. PKP2 has also been implicated in driving alterations to sodium channel current homeostasis and Nav1.5 localization or expression in ARVC25,26. Whole-cell patch-clamp electrophysiological analysis of adult PKP2 Hom cardiomyocytes revealed alterations in sodium current homeostasis compared to that of controls (Extended Data Fig. 2). Immunofluorescence microscopy also showed loss of Nav1.5 cell–cell junction and membrane protein localization in PKP2 Hom cardiomyocytes when compared with controls (Extended Data Fig. 2). Together, these data suggest that PKP2 Hom mice recapitulate key electrical disease features found to be characteristic of ARVC.
Structural defects are also observed in human ARVC; these are desmosomal ultrastructural deficits and fibrofatty replacement of the myocardium3,27. Transmission electron microscopy analysis of PKP2 Hom hearts at 4 weeks of age revealed enlarged gaps and an accumulation of multi-membrane vesicles at the cardiac cell–cell junction (Extended Data Fig. 3), indicative of protein degradation (protein degradation machinery accumulation) at this level. A similar phenomenon was observed in hearts deficient of constitutive photomorphogenesis 9 signalosome subunit 6 (CSN6), a desmosomal resident protein that, when lost, drives desmosomal protein degradation and ARVC28. Quantitative analysis of electron-dense desmosomes at the cell–cell junction revealed significantly reduced levels of desmosomes in PKP2 Hom hearts as early as 2 weeks of age compared to controls (Extended Data Fig. 3), suggesting early postnatal loss of desmosomes in PKP2 Hom hearts. To assess whether PKP2 Hom mice recapitulate fibrofatty replacement of myocardium, cardiac histological analysis was performed at 6 weeks of age. Hematoxylin and eosin staining revealed extensive dilation and thinning of both left and right ventricles in PKP2 Hom mice compared to littermate controls (Fig. 2a). Masson’s Trichrome staining highlighted severe loss of myocardium and fibrosis in both left and right ventricles (Fig. 2b,c). Quantitative PCR with reverse transcription (RT–qPCR) analysis validated the findings at the histological level, showing a significant increase in the expression of profibrotic genes Col1a1 and Col3a1 in PKP2 Hom mice compared to littermate controls (Fig. 2d). Studies in patients with ARVC and mouse models of ARVC have demonstrated a particular affinity for fat deposition to more specifically localize in the subepicardium of the right ventricle, which represents a critical location to understand the mechanisms of adipogenesis in ARVC24,29,30. Using Oil Red O staining as a marker for neutral lipids, we show that 6-week-old PKP2 Hom mouse hearts demonstrate lipid accumulation in the subepicardium of the right ventricle (Fig. 2e), a finding that was not observed in the left ventricles of PKP2 Hom mice and hearts of wild-type littermate control mice (Fig. 2e). Thus, PKP2 Hom mice display fibrofatty replacement of myocardium, a key hallmark of human ARVC. Recent evidence indicates that inflammatory pathways, such as cardiac NF-κB signaling, as well as secretory cytokine production are altered in ARVC settings31. We show that cardiac NF-κB protein expression is significantly upregulated in PKP2 Hom hearts compared to controls, and that cytokine array profile analyses reveal a significant upregulation of specific cardiac inflammatory cytokines in the serum of PKP2 Hom mice compared to controls (Extended Data Figs. 3 and 4). Together, these findings show the sufficiency of the PKP2 IVS10-1G>C mutation to recapitulate the postnatal onset of key human ARVC disease features in PKP2 Hom mice.

a,b, Representative cardiac sections stained with hematoxylin and eosin (a) and Masson’s Trichrome staining (b) from control and PKP2 Hom mice at 6 weeks of age. Scale bars, 1 mm. c, High-magnification views of Masson’s Trichrome stained sections from the left ventricle and right ventricle. Scale bars, 50 µm. d, Quantification of fibrosis with RT–qPCR for profibrotic gene markers Col1α1 and Col3α1 (controls, n = 5; Hom, n = 7 biologically independent animals). Data are presented as mean ± s.e.m. Two-tailed unpaired t-test. *P < 0.05 (P = 0.0116 for Col1α1 and P = 0.0448 for Col3α1). e, Right ventricle and left ventricle sections stained with Oil Red O from control and PKP2 Hom hearts at 6 weeks of age. Scale bars, 100 µm. Experiments were repeated independently three times with similar results.
PKP2 mutation affects cardiac PKP2 quality and quantity
To determine the molecular consequence of the PKP2 IVS10-1G>C mutation on PKP2 RNA, ventricular RNA was isolated from PKP2 Hom and littermate control hearts at 4 weeks of age. RT–qPCR analysis of mouse PKP2 exons 5–13 revealed a single, larger product in PKP2 Hom hearts at what appeared to be reduced levels of endogenous PKP2 in control hearts (Fig. 3a). A more focused RT–qPCR analysis of the mutation locus using PKP2 exons 9–10 primers similarly revealed a larger mutant product in PKP2 Hom hearts in the absence of an endogenous PKP2 product (Fig. 3a). RT–qPCR analysis of PKP2 exons 9–10 demonstrated a significant reduction in PKP2 transcript levels in PKP2 Hom hearts (Fig. 3a), suggesting that the PKP2 IVS10-1G>C splice acceptor site mutation affected total PKP2 RNA levels. Sanger sequencing analyses demonstrated contiguous sequences between PKP2 exons 9 and 10 in control hearts, suggestive of splicing using the canonical intron 9 splice acceptor site (Fig. 3b). However, sequencing analyses of PKP2 Hom hearts revealed an additional 54 base pairs between exons 9 and 10 (Fig. 3b), corresponding to the use of an alternative upstream splice site and suggestive of intron retention as a mechanism driving the larger PKP2 transcript size.

a, RT–qPCR analysis of PKP2 exons 5–13 and exons 9–10 in control and PKP2 Hom hearts at 4 weeks of age. RT–qPCR analysis of Pkp2 exons 9–10 in control and PKP2 Hom hearts (n = 4 biologically independent animals). Data are presented as mean ± s.e.m. Two-tailed unpaired t-test, **P < 0.01 (P = 0.0063). b, Sequencing analysis of RT–qPCR products from control and PKP2 Hom hearts. Red asterisk denotes mutation site. c, Western blot analysis of desmosomal (PKP2, DSP and DSG2), fascia adherens (JUP and N-Cad) and gap junction (CX43) proteins at 4 weeks of age in control and PKP2 Hom hearts. GAPDH served as the loading control. Endogenous WT and MUT PKP2 protein bands are depicted by black arrows. d, Quantification of protein expression in a normalized to GAPDH (n = 3 biologically independent animals). Experiments were repeated independently three times with similar results. Data are presented as mean ± s.e.m. Two-way ANOVA with Sidak’s multiple comparison test. ****P < 0.0001. e, Immunofluorescence staining of desmosomal, fascia adherens and gap junction proteins at 4 weeks of age in control and PKP2 Hom hearts. Scale bars, 25 µm. White arrows indicate the localization of mutant PKP2 in PKP2 Hom mice hearts. Experiments were repeated independently three times with similar results.
To determine the effect of the PKP2 IVS10-1G>C mutation on PKP2 and cardiac cell–cell junction protein homeostasis, western blot analysis was performed in ventricular lysates from PKP2 Hom hearts and littermate controls at 4 weeks of age. A higher molecular weight mutant PKP2 protein was identified in the absence of endogenous PKP2 in PKP2 Hom hearts (Fig. 3c). The mutant PKP2 protein was found at significantly reduced levels compared to wild-type PKP2 in littermate control hearts (Fig. 3c,d), suggesting the translation of the larger sized PKP2 transcript into a higher molecular weight mutant PKP2 protein (Fig. 3c). These molecular consequences resulted in the direct loss of neighboring desmosomal components, including DSP, DSG2 and JUP, in PKP2 Hom hearts (Fig. 3c,d). The molecular alterations at the desmosome had direct consequences on the predominant ventricular gap junction protein, CX43, which was significantly reduced in PKP2 Hom hearts compared to controls (Fig. 3c–e). CX43 is thought to be a direct target of DSP, suggesting a molecular consequence of desmosomal disruption24. At this time point, levels of the fascia adherens marker N-Cad in PKP2 Hom hearts were not significantly different from those of controls (Fig. 3c,d). Using the cell–cell junction marker N-Cad, immunofluorescence microscopy analyses showed that the mutant PKP2 as well as CX43, JUP and DSP were localized at the cell–cell junction in PKP2 Hom hearts, albeit at reduced levels compared to those in control hearts (Fig. 3e), which is consistent with findings from western blot analysis (Fig. 3c,d). Together, these data demonstrate that the PKP2 IVS10-1G>C splice acceptor site mutation affects PKP2 protein quality and quantity by the appearance of a higher molecular weight mutant PKP2 protein that seems to retain cell–cell junction localization, and by the loss of endogenous PKP2 protein, which results in reduced total PKP2 protein levels compared to littermate controls.
PKP2 quantity deficits drive desmosomal protein loss
To determine whether the early postnatal desmosomal disruption in PKP2 Hom hearts is driven by alterations in PKP2 quality (that is, the toxicity of mutant PKP2) or quantity (that is, reduced protein levels compared to wild-type PKP2), we generated adenovirus vectors expressing wild-type (Ad WT) and mutant (Ad MUT) mouse PKP2 to determine their effects on desmosomal protein levels in PKP2 Hom neonatal mouse cardiomyocytes (Fig. 4a). At postnatal day 1–2, PKP2 Hom mice do not show overt ARVC disease features, yet display ARVC-related molecular alterations, including disruption or loss of components of the desmosome (Extended Data Fig. 5). We observed a significant reduction in the desmosomal components PKP2 (endogenous), DSP, DSG2 and JUP, as well as appearance of a higher molecular weight PKP2 in uninfected PKP2 Hom neonatal cardiomyocytes compared to control cardiomyocytes (Fig. 4). However, Ad PKP2 WT-treated PKP2 Hom neonatal cardiomyocytes significantly increased DSP, DSG2 and JUP expression in PKP2 Hom neonatal cardiomyocytes compared to uninfected PKP2 Hom cardiomyocytes (Fig. 4). Interestingly, PKP2 Hom neonatal cardiomyocytes treated with Ad PKP2 MUT also demonstrated a significant increase in DSP, DSG2 and JUP protein expression compared to uninfected PKP2 Hom cardiomyocytes (Fig. 4). Given that the restoration of either wild-type or mutant PKP2 protein was sufficient to increase desmosomal protein levels, these data highlight that loss of PKP2 protein quantity and not quality (that is, mutant form) is the driver of desmosomal disruption in PKP2 Hom neonatal cardiomyocytes and early ARVC. Furthermore, these findings show the ability of PKP2 to function as a potent molecular scaffold to restore near-wild-type expression levels of proteins at the desmosomal cell–cell junction.

a, Representative western blot analysis of desmosomal (PKP2, DSP, DSG2, JUP) and fascia adherens junction (N-Cad) proteins in control and Hom cardiomyocytes infected with PKP2 WT and PKP2 MUT adenoviruses. Uninfected cells were used as a control. The experiments were repeated using five independent preparations of cells. Western blot analysis of GAPDH was used as a loading control. CM, cardiomyocyte. b, Quantification of western blot analysis. Data are presented as mean ± s.e.m., n = 3 individual experiments. One-way ANOVA with Tukey’s multiple comparison test was used to compare the significance among the treatments for the same protein. *P < 0.05 (PKP2, P = 0.0408), **P < 0.01 (DSG2, P = 0.0047), ***P < 0.001 (DSP, P = 0.0001; DSG2, P = 0.0009; JUP, P = 0.0004), ****P < 0.0001.
Early PKP2 restoration prevents ARVC deficits in mice
To determine whether restoring cardiac PKP2 protein levels can affect cardiac desmosomal protein disruption and disease in PKP2 Hom mice in vivo, we designed an AAV strategy to express wild-type mouse PKP2 (with carboxy-terminal tagged with FLAG; AAV-PKP2) in the heart using a cardiotropic AAV9 serotype and cardiac troponin T (cTnT) promoter (Fig. 5a). AAV9 tagged with green fluorescent protein (AAV-GFP) was used as a control. PKP2 Hom mice were injected with AAV at 5 × 1011 genome copies per mouse on postnatal day 2 and subsequently analyzed at 4 weeks of age (Fig. 5b), a time point at which all ARVC disease features are present (Figs. 1 and 2). The virus dose was based on previous studies using neonatal AAV injection in a mouse model of catecholaminergic polymorphic ventricular tachycardia32. Immunofluorescence analysis showed FLAG staining localized at the cardiomyocyte cell–cell junction in AAV-PKP2-treated PKP2 Hom hearts, as evidenced by colocalization with the desmosomal marker JUP within cardiomyocytes marked with alpha-actinin staining, compared to uninjected controls (Fig. 5c), demonstrating that exogenous PKP2 can localize to the cardiomyocyte cell–cell junction following AAV delivery. Western blot analyses revealed that neonatal administration of AAV-PKP2 was sufficient to restore PKP2 protein to endogenous levels in 4-week-old PKP2 Hom hearts (Fig. 5d,e). PKP2 restoration was also sufficient to significantly increase levels of other desmosomal components (DSP, DSG2 and JUP), as well as of the integral gap junction component CX43 (Fig. 5d,e) in 4-week-old PKP2 Hom hearts. The fascia adherens marker N-Cad remained unchanged at 4 weeks post AAV-PKP2 treatment, similar to untreated controls (Fig. 5d,e). These findings were in contrast to untreated PKP2 Hom mice, which demonstrated significant desmosomal (PKP2, DSP, DSG2, JUP) dissolution and gap junction (CX43) protein reduction (Fig. 5d,e). These data further highlight the selective ability of PKP2, as a single desmosomal gene, to scaffold and reassemble cardiac cell–cell junction components at and beyond the cardiac desmosome.

a, Design of AAV Pkp2 vector with inverted terminal repeats (ITR), cTnT promoter, Kozak sequence, Pkp2 cDNA, C-terminal FLAG tag, Woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) and bovine growth hormone polyadenylation signal (BGH-pA). b, Schemata of early neonatal delivery strategy and analysis time point. c, Representative immunofluorescence staining of FLAG (green), JUP (red) and alpha-actinin (white) at 4 weeks post injection in uninfected wild-type control and PKP2 Hom hearts treated with AAV-PKP2. Scale bars, 20 µm. Experiments were repeated independently three times with similar results. d, Western blot analysis of desmosomal, fascia adherens and gap junction proteins at 4 weeks post injection in uninfected control, uninfected PKP2 Hom and AAV-PKP2-treated PKP2 Hom hearts. GAPDH served as the loading control. e, Quantification of protein expression in (d) normalized to GAPDH (controls, n = 5; Hom, n = 5; Hom-AAV-PKP2, n = 7 biologically independent animals). Data are presented as mean ± s.e.m. Two-way ANOVA with Tukey’s multiple comparison test. ****P < 0.0001, ***P < 0.001 (DSG2, P = 0.0008), *P < 0.05 (CX43, Ctrl versus Hom-AAV-PKP2, P = 0.0123; Hom versus Hom-AAV-PKP2, P = 0.0174). Experiments were repeated independently three times with similar results. f, Hematoxylin and eosin staining of cardiac sections from wild-type control and PKP2 Hom mice treated with AAV-GFP or AAV-PKP2. Scale bars, 1 mm. g, Ratios of heart weight to body weight of wild-type control and PKP2 Hom mice treated with AAV-GFP or AAV-PKP2 (controls, n = 6; Hom, n = 4; Hom-AAV-PKP2, n = 8 biologically independent animals). Data are presented as mean ± s.e.m. One-way ANOVA with Tukey’s multiple comparison test. ***P < 0.001 (P = 0.0008), **P < 0.01 (P = 0.0014). h, Masson’s Trichrome staining of cardiac sections from wild-type control and PKP2 Hom mice treated with AAV-GFP or AAV-PKP2. Scale bars, 100 µm. i, RT–qPCR analysis of Col1α1 levels (controls, n = 4; Hom, n = 4; Hom-AAV-PKP2, n = 6 biologically independent animals). Data are presented as mean ± s.e.m. One-way ANOVA with Tukey’s multiple comparison test. *P < 0.05 (P = 0.0316 for Ctrl versus Hom; P = 0.0429 for Hom versus Hom-AAV-PKP2).
To determine the effect of AAV-PKP2 on cardiac morphology and remodeling in PKP2 Hom mice, we performed gross morphological and histological analyses. Hematoxylin and eosin staining of whole heart sections revealed that cardiac dimensions and cardiac muscle integrity were affected in PKP2 Hom mice treated with AAV-GFP, which exhibited enlarged left and right ventricular chambers and extensive hematoxylin (nuclear) staining in the left ventricle free wall (Fig. 5f), suggestive of cardiomyocyte necrosis and calcification. By contrast, PKP2 Hom mice treated with AAV-PKP2 were indistinguishable from wild-type controls, showing no evidence of disease (Fig. 5f). Ratios of heart weight to body weight revealed similar findings (Fig. 5g). However, PKP2 Hom hearts treated with AAV-GFP displayed significantly higher ratios of heart weight to body weight compared to controls and AAV-PKP2-treated PKP2 Hom mice (Fig. 5g). Masson’s Trichrome (collagen) stains revealed that cardiac sections from AAV-PKP2-treated PKP2 Hom mice were indistinguishable from those from wild-type controls, suggesting the absence of fibrosis (Fig. 5h). By contrast, extensive fibrotic areas were present in both left and right ventricles of PKP2 Hom mice treated with AAV-GFP (Fig. 5h). RT–qPCR for fibrotic gene expression (Col1a1) also revealed a prevention of fibrotic remodeling in PKP2 Hom mice treated with AAV-PKP2, as their Col1a1 levels were indistinguishable from those of controls (Fig. 5i). By contrast, PKP2 Hom mice treated with AAV-GFP exhibited a significant induction of fibrotic gene expression (Fig. 5i). Together, these data suggest that AAV-PKP2 can prevent pathological cardiac remodeling and fibrosis in 4-week-old PKP2 Hom mice.
To determine the functional and electrical effect of AAV-PKP2 in 4-week-old PKP2 Hom mice in vivo, we performed cardiac MRI and surface ECG analyses. Representative short-axis MRI views highlighted the positive effect of AAV-PKP2 on cardiac dimensions at end-diastole, particularly in the right ventricle (Fig. 6a). Quantitative MRI analysis revealed no significant differences in heart rates between groups; however, cardiac function, assessed by ejection fraction, was significantly improved in both left and right ventricles following AAV-PKP2 treatment compared to AAV-GFP treatment (Fig. 6b). This was accompanied by significant decreases in right ventricular end-diastolic and end-systolic volumes as well as left ventricular end-systolic volumes in the AAV-PKP2-treated mice compared to controls (Fig. 6b). The analysis of composite surface ECG tracings (Fig. 6c) revealed a significantly widened QRS complex in AAV-GFP-treated PKP2 Hom mice (Fig. 6d), which was significantly corrected to wild-type control values in PKP2 Hom mice treated with AAV-PKP2 (Fig. 6d). Furthermore, no observed differences in heart rate or PR interval were found between groups (Fig. 6d), highlighting a specific effect of AAV-PKP2 in alleviating ventricular depolarization delay. Surface ECG analyses also revealed that 60% of AAV-GFP-treated PKP2 Hom mice exhibited PVCs at 4 weeks of age (Fig. 6e,f), whereas 0% of wild-type control and 0% of PKP2 Hom mice treated with AAV-PKP2 showed PVCs (Fig. 6e,f). Together, these data demonstrate that AAV-PKP2 can alleviate the severe cardiac electrical and mechanical deficits found in PKP2 Hom mice.

a, Representative short-axis MRI from wild-type control mice, PKP2 Hom mice treated with AAV-GFP and PKP2 Hom mice treated with AAV-PKP2. b, Quantification of heart rate, ejection fraction, end-diastolic volume and end-systolic volume (controls, n = 5; Hom-AAV-GFP, n = 5; Hom-AAV-PKP2, n = 6 biologically independent animals). Data are presented as mean ± s.e.m. One-way ANOVA with Tukey’s multiple comparison test for heart rate measurement. Two-way ANOVA with Bonferroni’s multiple comparison test for additional measurements. Adjusted P values, ****P < 0.0001, **P < 0.01, *P < 0.05 (ESV, LV, Ctrl versus Hom-AAV-GFP, P = 0.0210; Hom-AAV-GFP versus Hom-AAV-PKP2, P = 0.0039). c, Representative composite surface ECG tracings averaged from four beats in wild-type control and PKP2 Hom mice treated with AAV-GFP or AAV-PKP2 at 4 weeks of age. Scale bar, 10 ms. d, Quantification of heart rate, PR interval and QRS interval from composite surface ECG tracings (controls, n = 5; Hom-AAV-GFP, n = 5; Hom-AAV-PKP2, n = 4 biologically independent animals). Data are presented as mean ± s.e.m. One-way ANOVA with Tukey’s multiple comparison test. *P < 0.05 (Ctrl versus Hom-AAV-GFP, P = 0.0149; Hom-AAV-GFP versus Hom-AAV-PKP2, P = 0.0348). e, Representative ECG tracings through time from control and PKP2 Hom mice treated with AAV-GFP or AAV-PKP2 at 4 weeks of age. f, Quantification of mice demonstrating PVCs (red arrows) (controls, n = 5; Hom-AAV-GFP, n = 5; Hom-AAV-PKP2, n = 4 biologically independent animals).
Early PKP2 restoration affords long-term ARVC protection
To determine the long-term effect of AAV-PKP2 treatment in PKP2 Hom mice in vivo, we assessed cardiac cell–cell junction protein homeostasis, function and survival in mice up to 6 months of age (Fig. 7a). Kaplan–Meier survival analysis demonstrated 100% survival of AAV-PKP2-treated PKP2 Hom mice at 6 months of age, which was indistinguishable from survival in wild-type littermate control mice (Fig. 7b). This finding is in stark contrast to results in untreated PKP2 Hom mice, which exhibited a median survival of 11 weeks, with 0% survival at 6 months of age (Fig. 7b). Given that no untreated or AAV-GFP-treated PKP2 Hom mouse survived to 6 months of age, data from AAV-PKP2-treated PKP2 Hom mice were compared to data from wild-type littermate controls or to historical data from untreated or AAV-GFP-treated PKP2 Hom mice at earlier disease stages as a reference. Western blot analysis revealed that AAV-PKP2 treatment prevented dissolution of the cardiac cell–cell junction in PKP2 Hom mice (Fig. 7c,d), which was apparent as early as postnatal day 1–2 in untreated PKP2 Hom mice (Extended Data Fig. 5). We found that PKP2 protein levels persisted in hearts from AAV-PKP2-treated PKP2 Hom mice at 6 months of age compared to hearts from littermate controls (Fig. 7c,d). Persistent PKP2 expression prevented the loss of desmosomal (DSP, DSG2 and JUP), fascia adherens (N-Cad) and gap junction (CX43) proteins in 6-month-old PKP2 Hom hearts treated with AAV-PKP2, as they were indistinguishable from littermate control hearts (Fig. 7c,d). By contrast, untreated PKP2 Hom mice displayed a complete dissolution of the cardiac cell–cell junction, as shown by the significant reduction in desmosomal, gap junction and fascia adherens junction protein levels at 8 weeks of age compared to those of wild-type littermate control hearts (Extended Data Fig. 6). Representative short-axis MRI views revealed similar cardiac morphology between wild-type littermate control and AAV-PKP2-treated PKP2 Hom mice (Fig. 7e). Quantification of cardiac function via ejection fraction highlighted no significant differences in left ventricular function between 6-month-old wild-type littermate controls and AAV-PKP2-treated PKP2 Hom mice, with AAV-PKP2 treatment eliciting near-wild-type control levels in terms of right ventricular function in PKP2 Hom mice (Fig. 7f). This finding is in marked contrast to that obtained in AAV-GFP-treated PKP2 Hom mice, which showed a significant reduction in both left and right ejection fraction at 6 weeks of age (Fig. 7f), highlighting that AAV-PKP2 slows rapid cardiac dilatation and deterioration of cardiac function in PKP2 Hom mice. An assessment of electrical function via composite surface ECG tracing analysis revealed that 6-month-old AAV-PKP2-treated PKP2 Hom mice exhibited similar heart rates and QRS intervals to wild-type littermate controls, including the absence of PVCs (Fig. 7g,h). These data suggest that early postnatal administration of AAV-PKP2 prevents the electrical deficits, including ventricular depolarization delay and cardiac arrhythmias, in PKP2 Hom mice in the long term (6 months). Given that the liver represents a major off-target site with AAV treatment, we assessed circulating levels of liver enzymes alkaline phosphatase (ALP) and alanine aminotransferase (ALT) as a measure of liver function. We observed no significant differences in the levels of either enzyme between wild-type littermate control and AAV-PKP2-treated PKP2 Hom mice at 6 months of age (Fig. 7i), suggesting that liver function is not affected by long-term PKP2 gene therapy. These data highlight that AAV-PKP2 can drive persistent PKP2 protein expression, which elicits long-term desmosomal protection to prevent severe electrical and mechanical cardiac dysfunction as well as premature lethality in PKP2 Hom mice.

a, Schemata for early injection of AAV-GFP or AAV-PKP2 to PKP2 Hom mice at postnatal day 2 and post analysis at 6 months. b, Kaplan–Meier survival analysis (log-rank test) of mice (controls, n = 18; Hom, n = 23; Hom-AAV-PKP2, n = 7 biologically independent animals). Ctrl and PKP2 Hom data in Fig. 7 are also presented in Fig. 1. ****P < 0.0001 (Χ2 58.07, d.f. 2). c, Western blot analysis of PKP2 and cell–cell junctional proteins (DSP, DSG2, JUP, N-Cad and CX43) in mouse hearts. GAPDH was used as a loading control. d, Quantification of protein expression shown in c normalized to GAPDH. Data are presented as mean ± s.e.m., n = 5 biologically independent animals. Two-way ANOVA with Sidak’s multiple comparison test. ****Adjusted P < 0.0001. e, Representative short-axis cardiac MRI views. f, Quantification of heart rate and ejection fraction in mice using cardiac MRI (controls, n = 4; Hom-AAV-GFP, n = 6; Hom-AAV-PKP2, n = 5 biologically independent animals). Data are presented as mean ± s.e.m. One-way ANOVA for heart rate comparison. Two-way ANOVA with Bonferroni’s multiple comparison test for other comparisons. Adjusted P values, ****P < 0.0001, *P < 0.05 (P = 0.0358). Historical data for Hom-AAV-GFP are from 6 weeks as no PKP2 Hom mouse survived to 6 months of age. g, Representative composite surface ECG tracings averaged from four beats in untreated wild-type control and PKP2 Hom mice treated with AAV-PKP2. Scale bar, 10 ms. h, Quantification of heart rate and QRS intervals from composite surface ECG tracings (controls, n = 5; Hom-AAV-PKP2, n = 6 biologically independent animals). Data are presented as mean ± s.e.m. Two-tailed unpaired t-test. NS, not significant. i, Representative ECG tracings from untreated control and PKP2 Hom mice treated with AAV-PKP2. j, Blood serum analysis for ALT and ALP liver enzyme levels in untreated wild-type control and PKP2 Hom mice treated with AAV-PKP2 (controls, n = 5; Hom-AAV-PKP2, n = 6 biologically independent animals). Normal enzyme limits are indicated with dotted lines. Data are presented as mean ± s.e.m. Two-tailed unpaired t-test.
Late PKP2 restoration rescues ARVC deficits in mice
To evaluate the effect of PKP2 gene therapy in the setting of existing ARVC, PKP2 Hom mice were injected with AAV at 5 × 1011 genome copies per mouse at 4 weeks of age (Fig. 8a), a time point at which all gross ARVC disease features are present (Figs. 1 and 2). Mice were euthanized 2 weeks (Fig. 8a) and 5 weeks (Extended Data Fig. 7) post AAV administration to assess early and longitudinally late ARVC molecular and functional outcomes. As early as 2 weeks post AAV injection, AAV-PKP2 treatment was sufficient to increase PKP2 levels and significantly improve desmosomal protein (DSP, DSG2 and JUP) levels in PKP2 Hom mice compared to those in AAV-GFP-treated PKP2 Hom mice (Fig. 8b). In addition, representative cardiac short-axis MRI views at both end-diastole and end-systole showed an improvement in cardiac morphology in AAV-PKP2-treated PKP2 Hom mice at 6 weeks of age compared to littermate AAV-GFP-treated PKP2 Hom mice (Fig. 8c). Quantitative MRI analysis revealed no significant differences in heart rates between mice, but it showed a significant improvement in left and right ventricular ejection fraction in AAV-PKP2-treated PKP2 Hom mice compared to AAV-GFP-treated PKP2 Hom mice (Fig. 8d). We also found that as late as 5 weeks post AAV injection, AAV-PKP2 treatment retained the ability to alleviate desmosomal protein loss, resulting in longitudinal effects on alleviating cardiac fibrofatty deposition and arrhythmias, as well as further improving cardiac right and left mechanical function in adult PKP2 Hom mice compared to AAV-GFP-treated controls (Extended Data Figs. 7 and 8). Immunofluorescence microscopy also revealed Nav1.5 protein relocalization to the cell–cell junction and membrane in AAV-PKP2-treated PKP2 Hom mice compared to AAV-GFP-treated controls (Extended Data Fig. 2). Furthermore, NF-κB protein expression and specific sets of cardiac inflammatory cytokines were also downregulated in hearts and serum of AAV-PKP2-treated PKP2 Hom mice, respectively, compared to AAV-GFP-treated controls (Extended Data Figs. 3 and 4). Survival analysis revealed 100% survival of AAV-PKP2-treated PKP2 Hom mice at 20 weeks of age compared to 20% survival of AAV-GFP-treated PKP2 Hom mice at the same age (Fig. 8e,f). Immunofluorescence microscopy of FLAG expression showed that this one-time late-stage administration of AAV-PKP2 (which was tagged with FLAG) at 5 weeks post AAV administration was sufficient to infect a majority of cardiomyocytes in the left and right ventricles of PKP2 Hom mice (Extended Data Fig. 9), concurrent with improvements in cardiac desmosomal biology, histopathology, rhythm and physiology in PKP2 Hom mice at these late stages. These results highlight the restorative actions and durability of AAV-PKP2 to rescue multiple parameters associated with ARVC deficits, which resulted in prolongation of lifespan of PKP2 Hom mice, even when administered in advanced stages of ARVC.

a, Schemata for late injection of AAV-GFP or AAV-PKP2 to PKP2 Hom mice at 4 weeks and post analysis at 6 weeks. gc, genom copies. b, Western blot analysis of PKP2 and desmosomal cell–cell junctional proteins (DSP, DSG2 and JUP) in mouse hearts. GAPDH was used as a loading control. Experiments were repeated independently three times with similar results. c, Representative short-axis MRI views from mice. d, Quantification of heart rate and ejection fraction in mice using cardiac MRI (n = 6 biologically independent animals). Data are presented as mean ± s.e.m. Two-way ANOVA with Sidak’s multiple comparison test. **P < 0.01 (P = 0.0026), *P < 0.05 (P = 0.0450). e, Schemata for late injection of AAV-GFP or AAV-PKP2 to PKP2 Hom mice at 4 weeks and post analysis at 20 weeks. gc, genom copies. f, Kaplan–Meier survival analysis (log-rank test) of mice (controls, n = 8; Hom-AAV-GFP, n = 8; Hom-AAV-PKP2, n = 7 biologically independent animals. ***adjusted P < 0.001 (Χ2 16.59, d.f. 2, P = 0.0003).
Discussion
In this study, we generate a PKP2 genetic knock-in mouse model to demonstrate the sufficiency of a prevalent PKP2 RNA splicing mutation (human PKP2 IVS10-1G>C) to phenocopy key human ARVC features in mouse, including sudden death, ventricular arrhythmias (which include ventricular depolarization abnormalities serving as a primer for life-threatening arrhythmias, as well as electrophysiological alterations in sodium channel function and Nav1.5 protein localization), biventricular dysfunction, inflammation, desmosomal ultrastructural deficits and fibrofatty replacement of myocardium. Although we acknowledge that more research is needed to fully understand the extent to which the observed changes in PKP2 Hom mice are present in humans, we believe our study represents an important step in unraveling complex mechanisms underlying PKP2-related ARVC, especially related to splicing mutations, providing a foundation for future investigations in both animal models and clinical settings. Furthermore, we show that early- and late-stage administration of PKP2 gene therapy can be used as a means to circumvent and rescue ARVC development and mortality in this genetic PKP2 model that harbors patient genetics. Our studies show that the PKP2 IVS10-1G>C splice site mutation has direct consequences on PKP2 RNA quality (that is, appearance of a larger, mutant PKP2) and levels (that is, reduced levels), suggestive of partial intron retention as a mechanism. Interestingly, this partial intron retention remains in frame and is successfully translated to a higher molecular weight mutant PKP2 protein at reduced levels of endogenous PKP2. Previous work suggests that intron retention mechanisms decrease transcript levels through a frameshift and the introduction of a premature termination codon (PTC)18. PTCs are known to trigger rapid mRNA degradation through a quality control mechanism termed ‘nonsense-mediated decay’, ultimately reducing mutant transcript levels to prevent translation of a potential toxic or gain-of-function protein17,33. Our data imply that mutant PKP2 transcripts in PKP2 Hom hearts are PTC-free, which suggests the possibility that non-PTC-associated mechanisms decrease PKP2 transcript levels due to altered splicing kinetics from a weaker alternative intronic splice acceptor site. Future studies will focus on understanding the precise mechanisms by which altered PKP2 splicing drives PKP2 RNA consequences in PKP2 Hom mice.
The unique PKP2 RNA and protein consequences of the PKP2 IVS10-1G>C splice site mutation confer postnatal viability and desmosomal deficits leading to ARVC disease features in PKP2 Hom mice. The presence of mutant PKP2 protein at reduced levels at the cardiac cell–cell junction in PKP2 Hom hearts in vivo, and the findings of in vitro expression studies using mutant PKP2 in PKP2 Hom neonatal cardiomyocytes reveal that mutant PKP2 is retained and functional at the cell–cell junction and capable of restoring desmosomal protein levels. This probably accounts for the viability of PKP2 Hom mice during embryonic development. This is in line with studies performed in global PKP2 knockout mice, which demonstrated an embryonic lethal phenotype34, suggesting that the global loss of endogenous PKP2 offers no survival advantage to mice. Our data may also help to explain survival in transgenic mice expressing truncated mutant PKP2 (S329X) and AAV-mediated expression of truncated mutant PKP2 (R735X)35,36, as these models retained the presence of endogenous PKP2, which probably conferred a survival advantage. Another example of a desmosomal variant resulting in mutant byproducts that confer an advantage in terms of survival and disease-free status was shown in a mouse model harboring a JUP variant associated with Naxos disease, which is an autosomal recessive form of ARVC37,38. Characterization of JUP-mutant mice revealed that they expressed a truncated mutant JUP protein at significantly reduced levels compared to controls38. More importantly, generation of a model that increased levels of the truncated JUP protein to endogenous levels was sufficient to prevent disease development, further suggesting that the mutant JUP protein could function equivalent to the endogenous JUP protein in the heart38. Our data suggest that the molecular consequences of the PKP2 IVS10-1G>C splice acceptor site mutation on PKP2 protein levels during early cardiac development may confer more pronounced ARVC-related cardiac phenotypes (that may not be fully compensated by the mutant PKP2 protein), as we observed pronounced baseline arrhythmias and fatty replacement in conjunction with cardiac mechanical and pathological dysfunction in the PKP2 Hom model compared to other models that ablate or express mutations in PKP2 in adult heart settings35,36,39. Importantly, our data add to the field and provide direct evidence that desmosomal protein disruption associated with the PKP2 splice site mutation is driven by a loss of function (dose or amount of PKP2) mechanism over time and the progressive nature of ARVC. This is in line with a growing number of human studies that suggest haploinsufficiency as a common mechanism that may underlie ARVC associated with various PKP2 variants, including insertion or deletion, nonsense and missense subtypes12,13. As human patients with ARVC are heterozygous for the PKP2 IVS10-1G>C mutation, our data also contribute to the growing evidence that a two-hit hypothesis (additional pathogenic stimulus) is central to elicit rapid ARVC pathogenesis by way of an additional genetic (a second desmosomal mutation or homozygous mutation) or physiological (for example, exercise stress) hit, especially in mouse settings8,24,40. This is further exemplified in PKP2 Het mutant mice, which exhibited a latent phenotype on survival and primarily exhibited arrhythmias in the absence of cardiomyopathy when assessed over the same and longer timeframes as PKP2 Hom mice. These electrophysiological abnormalities may probably account for premature lethality and sudden cardiac death in PKP2 Het mice at later ages. Furthermore, our previous studies also highlight the requirement for two human desmosomal mutations—this PKP2 RNA splice site (IVS10-1G>C) mutation and a DSP mutation; and thus, a compound heterozygous mutation—in order to elicit early and severe ARVC in a heterozygous setting in mouse in vivo28. Regardless, PKP2 Het mutant mice will provide a unique model to exacerbate and tease out the arrhythmogenic substrate and mechanisms underlying ARVC in the absence of cardiomyopathy in the future, which is also reminiscent of the early concealed phase of ARVC.
ARVC is an incurable genetic heart disease with significant mortality through sudden cardiac death and progressive heart failure. Studies in PKP2 Hom mice most importantly show broad applicability of therapeutic approaches to restore PKP2 RNA and protein levels (through PKP2 gene therapy) for PKP2 ARVC populations, including splice site mutations. In vivo-based AAV-PKP2 strategies were sufficient to restore PKP2 in the early stages of ARVC development in PKP2 Hom mice, which was sufficient to prevent not only the striking cardiac desmosomal dissolution but also gap junction deficits that together drive the long-term electrical (arrhythmias) and structural (cardiac pathology and biventricular dysfunction) cardiac deficits in ARVC in PKP2 Hom hearts. This was further exemplified in the longitudinal late-stage administration studies, which showed that AAV-PKP2 could revert multiple histopathological cardiac abnormalities (reduction in cardiac fibrosis, fat deposition, inflammation and cytokine production) and physiological cardiac abnormalities (improvements in left and right ventricular dimensions and function, alleviation of cardiac arrhythmias) in PKP2 Hom mice. Interestingly, longitudinal studies at 5 weeks post AAV-PKP2 administration showed further improvements in mechanical function in PKP2 Hom mice compared to 2 weeks post AAV-PKP2 administration, highlighting a normalization of mechanical dysfunction as a function of time. The alleviation of cardiac arrhythmias was also supported by Nav1.5 protein relocalization to cell–cell junctions in PKP2 Hom hearts; however, future studies focused on evaluating the biophysical properties of the sodium channel will be required to fully determine its importance in the rescue of the electrophysiological abnormalities associated with PKP2-related disease. Regardless, cardiac desmosomal restoration and improvement in mechanical function are critical factors in preventing arrhythmias and maintaining cardiac health, and therefore can contribute to circumventing abnormalities that could lead to arrhythmias.
These data further highlight the ability of PKP2 to reconstitute the desmosome postnatally and function as a complex scaffold. PKP2 has a selective affinity for desmosomal proteins and is known to interact with the cytoplasmic tails of the transmembrane desmosomal cadherins (DSG2 and DSC2), as well as the plakin protein DSP within the desmosomal complex9,41. JUP, similar to PKP2, is an armadillo protein thought to also be involved in linking the desmosomal cadherins and DSP9,41. The partial intron retention within the mutant PKP2 protein does not contain any critical structural domains and is located towards the C terminus of PKP2. The amino-terminal head domain is known to be essential for the interaction of PKP2 with desmosomal components42,43, therefore it is unlikely that the mutant PKP2 protein directly disrupts desmosomal interactions but can certainly lessen them, and therefore, loss of interactions are probably due to the loss of PKP2 protein dose. This concept is further exemplified in global PKP2 knockout mice34; PKP2 protein loss in this setting could recapitulate the key desmosomal protein deficits found in neonatal and adult PKP2 Hom hearts. Thus, our findings provide evidence for the importance of PKP2 quantity in maintaining normal levels of cell–cell junction components (mutant PKP2 may preserve interaction to a certain degree but may be destabilizing postnatally as lower levels of mutant protein are expressed), supporting the concept of a critical threshold for PKP2-mediated regulation of cell–cell junction protein expression. Future studies focused on dissecting the molecular interactions and regulatory functions of PKP2 in controlling cell–cell junction component expression and determining whether the role of PKP2 is solely structural or extends to the control of protein expression will provide deeper understanding of the complex role of PKP2 in the pathogenesis of ARVC. The most striking effect of PKP2 gene therapy in PKP2 Hom mice is ensuring 100% survival when administered in early and late stages of the disease. Although the mechanisms remain to be investigated in detail, one possible mechanism could relate to its interactions with the desmosomal resident protein CSN6, which is thought to interact with PKP2 and DSP and restrict desmosomal protein degradation, as well as circumvent ARVC and premature death in mice28, and thus improve desmosomal protein half-life over time. Therefore, future studies should focus on uncovering whether CSN6 functions are integral to the circumvention of ARVC deficits and premature lethality associated with PKP2 gene therapy in PKP2 Hom mice. Given that the desmosome represents a primary molecular trigger for the pathogenesis of ARVC, the ability to reassemble this complex is critical for ARVC-targeted therapies. This approach goes beyond current strategies that focus on symptomatic relief of electrical deficits (that is, arrhythmias), by focusing on alleviating the structural and functional deficits as well as premature death that underlie ARVC.
In summary, AAV-PKP2 therapy represents an effective approach for both preventing and stabilizing ARVC disease progression in PKP2 Hom mice. The use of a cardiotropic AAV serotype and cardiac-specific cTnT promoter enable durable PKP2 expression in the heart with no adverse effects on the liver. Further evaluation of additional tissue types is required before ultimate application of this approach in clinical settings. Optimization of dose (and potential of AAV antibodies) and capsid to determine maximum phenotypic improvement within a range that could be safely administered to human patients will be required to ensure the safety, efficacy and feasibility of gene therapy strategies for successful translation to patient populations with ARVC.
Methods
All research and animal procedures were in full compliance with the ethical guidelines of the Institutional Animal Care and Use Committee of the University of California San Diego, and carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH).
Generation of experimental animals
Mouse lines harboring heterozygous PKP2 IVS10-1G>C mutation were previously generated using CRISPR–Cas9-mediated methods and used for this study28,44. The mutation template used to generate PKP2 IVS10-1G>C Het mice is presented in Fig. 1. Genomic DNA was extracted from mouse tails, and genomic fragments at target sites were amplified by PCR and sequencing. Genotype-positive knock-in mice were backcrossed with C57BL/6J mice (The Jackson Laboratory) for at least three generations to minimize potential off-target effects. PKP2 IVS10-1G>C Het mice were crossed to generate controls, PKP2 IVS10-1G>C Het, and PKP2 IVS10-1G>C Hom mutant mice. Both male and female mice were used for all studies. Environmental controls were set to maintain a temperature of 21–23 °C with a relative humidity of 45–55%. Temperature and humidity were monitored and a 12 h–12 h light–dark cycle was maintained, alternating at 6:00 and 18:00.
Magnetic resonance imaging
In vivo cardiac MRI was performed on a 7T horizontal bore MR scanner (Bruker) as previously described28. In brief, a quadrature volume coil (Bruker) was used for radiofrequency signal transmission and a two-channel surface array coil (RAPID MRI) was used for reception of the RF signal. Cardiac CINE images were acquired with an IntraGate (Bruker) retrospective gated two-dimensional gradient echo pulse sequence (FLASH) with the following parameters: TE = 3.1 ms, TR = 5.6 ms, flip angle = 7°, 300–400 repetitions and 20 frames. A field of view of 2.0 × 1.5 cm and a data matrix of 256 × 192 were specified for a spatial resolution of 0.078 mm per pixel. Equatorial frames containing the largest and smallest chamber diameters were selected to define the end-diastolic and end-systolic times, respectively. For MRI image analyses, two-dimensional endocardial contours were manually segmented and slice volumes and/or ejection fractions calculated using freely available software (Segment CMR version 4.0 R12067, MEDVISO) for each heart at end-diastole and end-systole (both left and right ventricle). Volumes from continuous slices were summed to generate total chamber volumes at end-diastole and end-systole. Ejection fractions were averaged over all continuous slices.
Surface and telemetry ECG
Surface ECG was performed as previously described24. In brief, mice were anesthetized with 5% isoflurane (Piramal, 6679401710) for 15 s and maintained at 1.5% isoflurane during the procedure. Needle electrodes (30-gauge) were inserted subcutaneously into the right forearm and left leg. Electrical activity was recorded for 5 min. For analysis, composite ECG tracings were generated for 100 consecutive beats and wave parts manually identified to generate heart rates, PR intervals and QRS intervals. For PVC analysis, ectopic beats were identified over the entire 5-min recording. For telemetry ECG recording, ECG transmitters (DSI) were subcutaneously inserted into the back of mice. After 5 days post surgery, ECGs were recorded for 3 days. The number of PVCs was analyzed using LabChart version 8.0.8 (ADINSTRUMENTS).
Histological analysis
Mouse hearts were perfused in a relaxation buffer consisting of 300 mM KCl (Sigma-Aldrich, P9333) in PBS and fixed with 4% paraformaldehyde (ThermoFisher Scientific, J61899.AK). Fixed hearts were embedded in Tissue-Tek O.C.T. Compound (Sakura, 4583) or dehydrated and embedded in paraffin as previously described24. Sections of between 5 µm and 10 μm thickness were cut. Whole-heart (5 μm) paraffin sections were stained with hematoxylin and eosin (Sigma-Aldrich, HT1079-1SET) and Masson’s Trichrome (Sigma-Aldrich, HT15) stains according to the manufacturer’s instructions. Whole-heart (10 μm) cryosections were stained with Oil Red O (Sigma-Aldrich, O0625) according to the manufacturer’s instructions. Images were acquired with the Hamamatsu Nanozoomer 2.0 HT Slide Scanner.
RNA analysis
Total RNA was isolated from hearts using TRIZOL (Invitrogen) according to the manufacturer’s instructions. The first-strand cDNA was generated using PrimeScript RT Reagent Kit with gDNA Eraser (Takara). RT–qPCR was performed using primer sequences (Extended Data Table 1) obtained from Integrated DNA Technologies and with KOD Extreme Hot Start DNA Polymerase (Sigma-Aldrich, 71975-3). RT–qPCR was performed on heart cDNA using primer sequences (Extended Data Table 1) with Power SYBR Green PCR master mix (Applied Biosystems, 43-091-55) and using a Bio-Rad Mastercycler. All values were normalized to GAPDH mRNA levels as indicated. PCR products were sequenced (Eton Biosciences).
Protein analysis
Total protein extracts were isolated from cardiomyocytes and ventricles as previously described24. Immunodetection of desmoplakin (mouse, 1:1,000, Bio-Rad, 2722-504), desmoglein 2 (mouse, 1:1,000, Fitzgerald, 10R-D106a), plakophilin 2 C-terminal (mouse, 1:2,000, Fitzgerald, 10R-P130b), plakophilin 2 N-terminal (goat, 1:1,000, Lifespan Biosciences, LS-B9231), plakoglobin (goat, 1:1,000, Sigma-Aldrich, SAB2500802), N-cadherin (rabbit, 1:1,000, Abcam, ab76057), connexin 43 (rabbit, 1:8,000, Sigma-Aldrich, C6219), Nav1.5 (rabbit, 1:500, Alomone Labs, ASC-005), NF-κB (rabbit, 1:500, Santa Cruz Biotechnology, sc-372), β-actin (mouse, 1:2,000, Santa Cruz Biotechnology, sc-47778) and glyceraldehyde 3-phosphate dehydrogenase (mouse, 1:2,000, Santa Cruz Biotechnology, sc-32233) was done as previously described24. Briefly, secondary antibodies were used based on antibody species and included donkey anti-rabbit IgG (H+L) HRP (1:100, ThermoFisher Scientific, A16023), donkey anti-mouse IgG (H+L) HRP (1:100, ThermoFisher Scientific, A16011) and donkey anti-goat IgG (H+L) HRP (1:100, ThermoFisher Scientific, A15999) according to the manufacturer’s instructions.
Immunofluorescence microscopy
Heart cryosections were fixed in 100% acetone (Fisher Scientific, A18-1) at −20 °C for 8 min and were blocked in 5% donkey serum (Sigma-Aldrich, D9663) or PBS (Gibco, 10010023) before incubation with antibodies. Sections were subsequently stained with primary antibodies against PKP2 (mouse, 1:100, Fitzgerald, 10R-P130b), desmoplakin (mouse, 1:100, Bio-Rad, 2722-504), plakoglobin (goat, 1:100, Sigma-Aldrich, SAB2500802), connexin 43 (rabbit, 1:100, Sigma-Aldrich, C6219), N-cadherin (rabbit, 1:100, Abcam, C6219), Nav1.5 (rabbit, 1:100, Alomone labs, ASC-005), FLAG (mouse, 1:100, Sigma-Aldrich, F3165), perilipin (rabbit, 1:100, Cell Signaling Technology, 3470S), sarcomere alpha actinin (mouse, Sigma-Aldrich, A7811), sarcomere alpha actinin (rabbit, abcam, ab68167) and secondary antibodies, such as donkey anti-mouse IgG (H+L) DyLight 488 (1:400, ThermoFisher Scientific, SA5-10166), donkey anti-mouse IgG (H+L) Alexa Fluor 555 (1:400, ThermoFisher Scientific, A31570), donkey anti-rabbit IgG (H+L) Alexa Fluor 488 (1:400, ThermoFischer Scientific, A21206), donkey anti-rabbit IgG (H+L) Alexa Fluor 568 (1:400, ThermoFisher Scientific, A10042) and donkey anti-goat IgG (H+L) Alexa Fluor 647 (1:400, ThermoFischer Scientific, A21447). Immunofluorescence images were acquired using a Leica SP8 confocal microscope.
Neonatal cardiomyocyte isolation
Ventricular cardiomyocytes were isolated from neonatal (1–2 days old) mouse hearts using collagenase type II (Worthington Biochemical Corporation, LS004174) or trypsin (Sigma-Aldrich, T4799) digestion methods and plated on laminin (Gibco, 23-017-015) as previously described24. Cardiomyocytes were subsequently infected with adenoviruses expressing either wild-type PKP2 (MOI 1.0) or mutant PKP2 (MOI 1.0) for 24 h and subsequently maintained in media consisting of DMEM (Life Technologies, 11330-032), M199 (Corning, 10-060), 5% FBS (Gibco, 16000069), 10% horse serum (Gibco, 16050122) and 1% penicillin-streptomycin-glutamine (ThermoFisher Scientific, 10378016). For protein analyses, cardiomyocytes were collected at 5 days post infection.
Adenovirus and adeno-associated virus vectors
Adenovirus vectors expressing either wild-type or mutant PKP2 were designed and synthesized containing a cytomegalovirus promoter, C-terminal PKP2 FLAG tag, and P2A-linked GFP (Vector Builder). Adenovirus was packaged and grown through the University of California San Diego viral vector core as previously described24. AAV vectors expressing wild-type PKP2 were designed and synthesized containing a cTnT promoter and C-terminal PKP2 FLAG tag (Vector Builder). In addition, AAV vector expressing GFP was designed and synthesized as a control. Viral particles were packaged into a cardiotropic AAV9 serotype and grown through the University of California San Diego viral vector core as previously described45.
Adeno-associated virus injections
Early AAV injections were performed on postnatal day 2 using a 31-gauge needle and syringe (Monoject, 8881600800) to deliver 5 × 1011 viral particles per mouse in 50 µl of solution containing AAV9-GFP or AAV9-PKP2 via intraperitoneal injection. Late-stage AAV injections were performed at 4 weeks of age using a 31-gauge needle and syringe to deliver 5 × 1011 viral particles per mouse in 50 µl of solution containing AAV9-GFP or AAV9-PKP2 via retro-orbital injection.
Whole-cell patch-clamp analysis
Whole-cell patch-clamp analysis of adult cardiomyocytes was performed as previously described, with some modifications46,47. In brief, single ventricular myocytes were isolated from mouse ventricles using the enzymatic digestion method by Langendorff. The single-pipette, whole-cell, voltage-clamp technique was performed by using a patch clamp amplifier (Axopatch 200B, Axon) to record membrane currents, and all experiments were performed at a room temperature of approximately 22 °C. The sodium channel current was recorded by stepped voltage-clamp (200 ms) between −80 and 15 mV in 5-mV steps from a holding potential of −80 mV, generated by a digital-to-analog converter (DigiData 1440, Axon) controlled by pCLAMP software (version 10.3, Axon). The modified Tyrode solution for perfusion contained 20 mM of NaCl (Sigma-Aldrich, 7683), 1 mM of MgCl2 (Sigma-Aldrich, M8266), 1 mM of CaCl2 (Sigma-Aldrich, C5670), 0.1 mM of CdCl2 (Sigma-Aldrich, 655198), 117.5 mM of CsCl (Sigma-Aldrich, 20966), 20 mM of HEPES (Sigma-Aldrich, 54457), 11 mM of glucose (Sigma-Aldrich, 7528) and 2 mM of nisoldipine (Sigma-Aldrich, N0165) with a pH of 7.4. The pipette solution contained 110 mM of CsCl (Sigma-Aldrich, 20966), 20 mM of EGTA (Sigma-Aldrich, 324626), 10 mM of TEA (Sigma-Aldrich, 86614), 5 mM of MgATP (Sigma-Aldrich, A9187) and 5 mM of HEPES (Sigma-Aldrich, 54457) with a pH of 7.2. Junction potentials were zeroed before formation of the membrane–pipette seal. Several minutes after seal formation, the membrane was ruptured by gentle suction to establish the whole-cell configuration for voltage clamping. Cell capacitance was measured by integrating the capacitive transient evoked by applying a 5-mV depolarizing step from a holding potential of −80 mV. Pipettes (resistance 3–5 MΩ) were pulled by a micropipette puller (Model P-87, Sutter Instrument).
Mouse serum analysis assays
For liver enzyme analysis, serum was collected from mice at 6 months post AAV-PKP2 treatment, and serum ALP and ALT levels were assessed using a VetScan2 chemistry analyzer (Zoetis). For cytokine analysis, mouse serum was collected from late-stage AAV-administration mouse cohorts and subjected to cytokine profile assays as previously described31 and following the manufacturer’s instructions (R&D Systems, ARY028). In brief, cytokine array membranes were blocked for 1 h at room temperature and subjected to overnight incubation at 4 °C with 200 µl of mouse serum, followed by antibody detection cocktail incubation at room temperature for 1 h and then streptavidin–horseradish peroxidase (1:2,000, ThermoFisher Scientific, A16011) incubation for 30 min at room temperature. Cytokine signals on arrays were detected using the Chemiluminescence Reagent Mix according to the manufacturer’s instructions.
Statistical analysis
All data are presented as mean ± s.e.m. GraphPad Prism was used for analyses, and significance was evaluated using Student’s two tailed t-test and one-way or two-way analysis of variance (ANOVA) with Tukey’s post-hoc multiple comparison tests. For Kaplan–Meier survival analysis, significance was assessed by the log-rank test. Categorical data was analyzed using Fisher’s exact test in RStudio software. A P value of <0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings in this study are available within the paper and associated files. Source data are provided with this manuscript.
References
Thiene, G., Corrado, D. & Basso, C. Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet J. Rare Dis. 2, 45 (2007).
Sen-Chowdhry, S. et al. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu. Rev. Med. 61, 233–253 (2010).
Mattesi, G. et al. Natural history of arrhythmogenic cardiomyopathy. J. Clin. Med. 9, 878 (2020).
Peters, S., Trummel, M. & Meyners, W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 97, 499–501 (2004).
Elias Neto, J. et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) - what we have learned after 40 years of the diagnosis of this clinical entity. Arq. Bras. Cardiol. 112, 91–103 (2019).
Idris, A., Shah, S. R. & Park, K. Right ventricular dysplasia: management and treatment in light of current evidence. J. Community Hosp. Intern. Med. Perspect. 8, 101–106 (2018).
Wang, W., James, C. A. & Calkins, H. Diagnostic and therapeutic strategies for arrhythmogenic right ventricular dysplasia/cardiomyopathy patient. Europace 21, 9–21 (2019).
Marcus, F. I., Edson, S. & Towbin, J. A. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J. Am. Coll. Cardiol. 61, 1945–1948 (2013).
Sheikh, F., Ross, R. S. & Chen, J. Cell-cell connection to cardiac disease. Trends Cardiovasc. Med. 19, 182–190 (2009).
Rampazzo, A. et al. Intercalated discs and arrhythmogenic cardiomyopathy. Circ. Cardiovasc. Genet. 7, 930–940 (2014).
Gerull, B. et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 36, 1162–1164 (2004).
Kirchner, F. et al. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ. Cardiovasc. Genet. 5, 400–411 (2012).
Rasmussen, T. B. et al. Truncating plakophilin-2 mutations in arrhythmogenic cardiomyopathy are associated with protein haploinsufficiency in both myocardium and epidermis. Circ. Cardiovasc. Genet. 7, 230–240 (2014).
Akdis, D. et al. Myocardial expression profiles of candidate molecules in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia compared to those with dilated cardiomyopathy and healthy controls. Heart Rhythm 13, 731–741 (2016).
Groeneweg, J. A. et al. Functional assessment of potential splice site variants in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Heart Rhythm 11, 2010–2017 (2014).
Lim, K. H. et al. Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc. Natl Acad. Sci. USA 108, 11093–11098 (2011).
Anna, A. & Monika, G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J. Appl. Genet. 59, 253–268 (2018).
Attali, R. et al. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum. Mol. Genet. 18, 3462–3469 (2009).
Guernsey, D. L. et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. PLoS Genet. 6, e1001081 (2010).
Watanabe, T. et al. A mutant mRNA expression in an endomyocardial biopsy sample obtained from a patient with a cardiac variant of Fabry disease caused by a novel acceptor splice site mutation in the invariant AG of intron 5 of the alpha-galactosidase A gene. Intern. Med. 52, 777–780 (2013).
Svensson, A. et al. Arrhythmogenic right ventricular cardiomyopathy - 4 Swedish families with an associated PKP2 c.2146-1G>C variant. Am. J. Cardiovasc. Dis. 6, 55–65 (2016).
Syrris, P. et al. Clinical expression of plakophilin-2 mutations in familial arrhythmogenic right ventricular cardiomyopathy. Circulation 113, 356–364 (2006).
Walsh, R. et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203 (2017).
Lyon, R. C. et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 23, 1134–1150 (2014).
Asimaki, A., Kleber, A. G. & Saffitz, J. E. Pathogenesis of arrhythmogenic cardiomyopathy. Can. J. Cardiol. 31, 1313–1324 (2015).
Cerrone, M. & Delmar, M. Desmosomes and the sodium channel complex: implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 24, 184–190 (2014).
Basso, C. et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur. Heart J. 27, 1847–1854 (2006).
Liang, Y. et al. Desmosomal COP9 regulates proteome degradation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Clin. Invest. 131, e137689 (2021).
Basso, C. & Thiene, G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat?. Cardiovasc. Pathol. 14, 37–41 (2005).
Te Riele, A. S. et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J. Cardiovasc. Electrophysiol. 24, 1311–1320 (2013).
Chelko, S. P. et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 140, 1491–1505 (2019).
Bezzerides, V. J. et al. Gene therapy for catecholaminergic polymorphic ventricular tachycardia by inhibition of Ca(2+)/calmodulin-dependent kinase II. Circulation 140, 405–419 (2019).
Lambert, J. M. et al. Mechanisms and regulation of nonsense-mediated mRNA decay and nonsense-associated altered splicing in lymphocytes. Int. J. Mol. Sci. 21, 1335 (2020).
Grossmann, K. S. et al. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J. Cell Biol. 167, 149–160 (2004).
Moncayo-Arlandi, J. et al. Molecular disturbance underlies to arrhythmogenic cardiomyopathy induced by transgene content, age and exercise in a truncated PKP2 mouse model. Hum. Mol. Genet. 25, 3676–3688 (2016).
Cruz, F. M. et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J. Am. Coll. Cardiol. 65, 1438–1450 (2015).
McKoy, G. et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355, 2119–2124 (2000).
Zhang, Z. et al. Normalization of Naxos plakoglobin levels restores cardiac function in mice. J. Clin. Invest. 125, 1708–1712 (2015).
Cerrone, M. et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 8, 106 (2017).
Delmar, M. & McKenna, W. J. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ. Res. 107, 700–714 (2010).
Lyon, R. C. et al. Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 116, 1462–1476 (2015).
Chen, X. et al. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta-catenin signaling. J. Biol. Chem. 277, 10512–10522 (2002).
Bass-Zubek, A. E. et al. Plakophilins: multifunctional scaffolds for adhesion and signaling. Curr. Opin. Cell Biol. 21, 708–716 (2009).
Ma, X. et al. CRISPR/Cas9-mediated gene manipulation to create single-amino-acid-substituted and floxed mice with a cloning-free method. Sci. Rep. 7, 42244 (2017).
Bravo-Hernandez, M. et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat. Med. 26, 118–130 (2020).
Chiang, C. E., Wang, T. M. & Luk, H. N. Inhibition of L-type Ca2+ current in guinea pig ventricular myocytes by cisapride. J. Biomed. Sci. 11, 303–314 (2004).
Sato, P. Y. et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 105, 523–526 (2009).
Acknowledgements
We are grateful for support from the Neuroscience Microscopy Shared Facility and Vector Development Core Laboratory at the University of California San Diego. F.S. received funding from the NIH (grants HL142251 and HL162369) and LEXEO Therapeutics. A.M. received funding from the NIH (grant HL121754), Additional Ventures and the Joe and Clara Tsai Foundation. The University of California San Diego Neuroscience Microscopy Shared Facility is supported by a grant from the NIH (P30 NS047101).
Author information
Authors and Affiliations
Contributions
F.S. conceived, designed and supervised experiments. F.S., W.H.B. and J.Z. wrote the manuscript. F.S., W.H.B., J.Z., Y.L., J.W., K.L.P. and A.M. revised the manuscript. W.H.B. and Y.L. characterized PKP2 mutant mice. W.H.B. and J.Z. designed, analyzed and performed cardiac AAV studies. W.H.B., J.Z. and E.J.G.-L. performed cardiac MRI analyses. W.H.B., J.Z. and Y.G. performed AAV injections. T.W. and L.N. performed electrophysiological analysis. A.D. and J.W. performed and analyzed PKP2 Hom neonatal cardiomyocytes studies. N.M. performed and analyzed PKP2 Hom neonatal heart studies. K.L.P. and A.M. provided advice and supervised cardiac AAV and electrophysiological studies.
Corresponding author
Ethics declarations
Competing interests
F.S. was a co-founder of Stelios Therapeutics (acquired by LEXEO Therapeutics) and is a co-founder and shareholder of Papillon Therapeutics Inc as well as a consultant and shareholder of LEXEO Therapeutics Inc. A.M. is a co-founder of and equity holder in Insilicomed Inc and Vektor Medical Inc. For A.M., findings reported in this study did not involve the companies he is involved with in any way, and these findings have no relationship with the business activities or scientific interests of either company. All other authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks Silvia Priori, Jens Waschke and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editor: Elvira Forte, in collaboration with the Nature Cardiovascular Research team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Telemetry electrocardiogram (ECG) and cardiac magnetic resonance image (MRI) analysis in PKP2 Het mice.
(a) Representative telemetry ECG tracings and premature ventricular contraction (PVC) counts from Ctrl and PKP2 heterozygous (Het) mice at 3 months of age. Red arrows indicate premature ventricular contractions. Scale bar = 0.1 s. Data are presented as mean ± S.E.M. n = 3 biologically independent animals examined per group, two-tailed unpaired t test, * p < 0.05 (p = 0.0135). (b) Quantification of end-diastolic volumes (EDV), end-systolic volumes (ESV) and percentage of ejection fraction (EF %) in WT and PKP2 Het mice at 10 months old of age using cardiac MRI. n = 4 biologically independent animals examined per group. Data are presented as mean ± S.E.M. Two-way ANOVA with Tukey’s multiple comparison test.
Extended Data Fig. 2 PKP2 Hom mice display various electrophysiological abnormalities that precede premature death.
(a) Representative telemetry ECG tracings from Ctrl and PKP2 Hom (Hom) mice. Note that PKP2 Hom mice display isolated PVCs, that transition to ventricular tachycardia and fibrillation. Red arrow indicates PVC; Blue arrows indicate ventricular tachycardia. Scale bar = 0.1 s. (b) Immunofluorescence staining of Nav1.5, plakoglobin (JUP), and the cardiomyocyte marker, sarcomeric alpha-actinin in representative mouse heart sections from 9 week old Ctrl, PKP2 Hom + AAV-GFP (five weeks post-late stage administration) and PKP2 Hom+AAV-PKP2 (five weeks post-late stage administration). Scale bar, 10 μm. White arrows depict Nav1.5 re-localization to cell-cell junction/membrane in PKP2 Hom hearts. Experiments were repeated independently three times with similar results. (c) Representative sodium current recording (left) and ion current-voltage (I-Vm) relationship of sodium current (right) in isolated adult cardiomyocytes from 8-9 week old Ctrl and PKP2 Hom mice, highlighting loss of sodium channel homeostasis in PKP2 Hom cardiomyocytes. Current amplitude has been normalized against cell capacitance (pA/pF). Data are presented as mean ± S.E.M. Ctrl, n = 6 biologically independent animals; PKP2 Hom, n = 8 biologically independent animals. Two-way ANOVA, **** p < 0.0001.
Extended Data Fig. 3 PKP2 Hom mice display cardiac ultrastructural and inflammatory deficits.
(a) Representative transmission electron micrographs from Ctrl and PKP2 Hom hearts at 4 weeks of age. White arrows denote healthy desmosomes. Quantification of desmosomes per μm of membrane in Ctrl and PKP2 Hom hearts at two weeks of age, denoted early loss of desmosomes in PKP2 Hom. Red arrows denote multi-membrane vesicles, suggestive of desmosomal structure degradation. Scale bar = 1 µm. Data are presented as mean ± S.E.M. two-tailed t test, n = 7 for Ctrl, n = 8 for PKP2 Hom biologically independent animals per group, respectively. ***, p < 0.001 (p = 0.0005). (b) Representative western blot and quantification analysis of nuclear factor-kappa B (NF-κB) expression in heart lysates from 9 week old Ctrl, PKP2 Hom + AAV-GFP (five weeks post-late stage administration) and PKP2 Hom+AAV-PKP2 (five weeks post-late stage administration) mice. β-actin used as a loading control. Data are presented as mean ± S.E.M. One-way ANOVA with Turkey’s multiple comparisons test, n = 3 biologically independent animals per group, **, p < 0.01 (p = 0.0099), ***, p < 0.001 (p = 0.0009).
Extended Data Fig. 4 PKP2 Hom mice display blood serum cytokine alterations that can be alleviated with late-stage administration of AAV PKP2.
Representative cytokine arrays are shown from serum collected from 9 weeks old Ctrl, PKP2 Hom + AAV-GFP (five weeks post-late stage administration) and PKP2 Hom+AAV-PKP2 (five weeks post-late stage administration) mice. Spots in red refer to reference markers to compare overall exposure levels. Spots in blue refer to cytokines (CXCL11) and C5/C5A (Complement component 5a) that are not significantly different between groups. Spots in green refer to cytokines (BLC (CXCL13/BCA-1); Eotaxin (CCL11); TIMP-1) that are altered in PKP2 Hom treated with AAV GFP and rescued by AAV PKP2 treatment in PKP2 Hom mice. Blood serum samples from Ctrl (n = 4 biologically independent animals), Hom-AAV GFP (n = 5 biologically independent animals) and Hom-AAV PKP2 (n = 6 biologically independent animals) were pooled for the cytokines array analysis.
Extended Data Fig. 5 PKP2 Hom neonatal (postnatal day 1, P1) hearts display desmosomal deficits in the absence of gross morphological changes to the heart.
(a) P1 heart weight to body weight ratios (HW/BW). Data are presented as mean ± S.E.M. n = 16 for Ctrl, n = 10 for Hom biologically independent animals, two-tailed unpaired t test, p = 0.2129, not significant. (b) RT-qPCR of cardiac stress markers atrial natriuretic factor (Nppa) and B-type natriuretic factor (Nppb) as well as (c) profibrotic genes collagen α1 types I (Col1α1) and III (Col3 α1) in P1 hearts. Data are presented as mean ± S.E.M. For (b) and (c), n = 3 biologically independent animals per group, two-tailed unpaired t test between groups, p = 0.4171 (Nppa), p = 0.9368 (Nppb), p = 0.2800 (Col1 α 1), p = 0.9655 (Col3 α 1), not significant. (d) Western blot analysis of desmosomal (PKP2, DSP, DSG2, JUP) and fascia adherens junction (JUP, N-Cad) proteins in neonatal cardiomyocytes from Ctrl and PKP2 Hom (P1) hearts. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) serves as the loading control. (e) Quantification of protein expression in (d) normalized to GAPDH (n = 3 biologically independent animals per group). Data are presented as mean ± S.E.M. Two-way ANOVA with Tukey’s multiple comparison test. ****, p < 0.0001. **, p < 0.01 (p = 0.0011). *, p < 0.05 (p = 0.0335).
Extended Data Fig. 6 Late-stage dissolution of the cardiac cell-cell junction in PKP2 Hom hearts.
(a) Western blot analysis of desmosomal, fascia adherens, and gap junction proteins at eight weeks of age in Ctrl and PKP2 Hom hearts. GAPDH serves as the loading control. (b) Quantification of protein expression in (a) normalized to GAPDH (n = 3 biologically independent animals per group). Data are presented as mean ± S.E.M. Two-way ANOVA with Tukey’s multiple comparison test. ****, p < 0.0001, ***, p < 0.001 (p = 0.0008), **, p < 0.01 (p = 0.0019).
Extended Data Fig. 7 Longitudinal assessment of late-stage administration of AAV PKP2 reveals continued effects on alleviating cardiac desmosomal deficits in addition to reduction in cardiac histopathology (fibrosis), arrhythmias and dysfunction at five weeks post-AAV administration.
(a) Schemata for late injection of AAV GFP or AAV PKP2 to PKP2 Hom mice at 4 weeks and post-analysis at 9 weeks (top). Western blot analysis of PKP2 and desmosomal proteins (DSP, DSG2 and JUP) in mouse hearts. β-actin used as a loading control. Data are presented as mean ± S.E.M. n = 6 biologically independent animals per group, Two-way ANOVA with Tukey’s multiple comparison test. *, p < 0.05 (PKP2, p = 0.0410), **, p < 0.01 (DSG2, p = 0.0099; JUP, p = 0.0020). (b) Representative Masson’s Trichrome stains from left (LV) and right ventricular (RV) sections from 9-week-old Ctrl, PKP2 Hom-AAV GFP (five weeks post-late stage administration) and PKP2 Hom-AAV PKP2 (five weeks post-late stage administration) mice. Scale bar, 2 mm. Experiments were repeated independently three times with similar results. (c) Quantification of mice demonstrating PVCs and sudden death at 9 weeks of age in Ctrl, PKP2 Hom-AAV GFP (five weeks post-late stage administration) and PKP2 Hom-AAV PKP2 (five weeks post-late stage administration) mice. (d) Quantification of end-diastolic volumes (EDV), end-systolic volumes (ESV) and percentage of ejection fraction (EF %) in 9 week old Ctrl, PKP2 Hom-AAV GFP (five weeks post-late stage administration) and Hom-AAV PKP2 (five weeks post-late stage administration) mice using cardiac MRI. Data are presented as mean ± S.E.M, n = 4 for Ctrl, n = 5 for Hom-AAV GFP group and n = 6 for Hom-AAV PKP2 biologically independent animals. Two-way ANOVA with Tukey’s multiple comparison test. ****, adjusted p < 0.0001. ***, p < 0.001 (EF, LV, p = 0.0008; RV, p = 0.0008). *, p < 0.05 (EDV, LV, p = 0.0369; RV, p = 0.0148; ESV, LV, p = 0.0352), ns, not significant.
Extended Data Fig. 8 PKP2 Hom mice display fat deposition that can be alleviated with late-stage administration of AAV PKP2.
Immunofluorescence microscopy analysis of perilipin (adipocyte marker) expression and localization in representative cardiac sections from 9 week old WT as well as PKP2 Hom mice at five weeks post-late stage administration of AAV GFP or AAV PKP2. White arrows denote increased number of positive perilipin stains in PKP2 Hom mouse hearts treated with AAV GFP and significant reduction in perilipin positive stained regions in PKP2 Hom mouse hearts treated with AAV PKP2. Scale bar = 30 μm. Experiments were repeated independently three times with similar results.
Extended Data Fig. 9 PKP2 Hom mice display AAV PKP2-FLAG expression and distribution in a majority of cardiomyocytes throughout the right and left ventricles.
Immunofluorescence microscopy analysis of FLAG expression and localization in representative cardiac sections from 9 week old PKP2 Hom mice at five weeks post-late stage administration of AAV PKP2. Scale bar = 500 μm. Two insets on the left represent high magnification views of cardiomyocytes from left ventricle. Two insets on the right represent high magnification views of cardiomyocytes from right ventricle. Please note the localization of FLAG signal to the cell-cell junction in the inset panels. Inset scale bar, 100 μm. Experiments were repeated independently three times with similar results.
Supplementary information
Supplementary Information
Supplementary Table 1
Source data
Source Data Fig. 3
Unprocessed western blots
Source Data Fig. 4
Unprocessed western blots
Source Data Fig. 5
Unprocessed western blots
Source Data Fig. 7
Unprocessed western blots
Source Data Fig. 8
Unprocessed western blots
Source Data Extended Data Fig. 3
Unprocessed western blots
Source Data Extended Data Fig. 5
Unprocessed western blots
Source Data Extended Data Fig. 6
Unprocessed western blots
Source Data Extended Data Fig. 7
Unprocessed western blots
Source_Data_Fig1
Numerical Source Data
Source_Data_Fig6
Numerical Source Data
Source _Data_Fig7
Numerical Source Data
Source_Data_Fig8
Numerical Source Data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bradford, W.H., Zhang, J., Gutierrez-Lara, E.J. et al. Plakophilin 2 gene therapy prevents and rescues arrhythmogenic right ventricular cardiomyopathy in a mouse model harboring patient genetics. Nat Cardiovasc Res 2, 1246–1261 (2023). https://doi.org/10.1038/s44161-023-00370-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44161-023-00370-3
This article is cited by
-
AAV9:PKP2 improves heart function and survival in a Pkp2-deficient mouse model of arrhythmogenic right ventricular cardiomyopathy
Communications Medicine (2024)
-
Restoring PKP2 in arrhythmogenic cardiomyopathy
Nature Cardiovascular Research (2023)
