
Abstract
Familial hypercholesterolemia (FH) is a genetic disorder mainly caused by defects in the low-density lipoprotein receptor (LDLR) gene, although it can also be due to alterations in the gene encoding apolipoprotein B (familial defective apoB or FDB) or in other unidentified genes. In Morocco, the molecular basis of FH is unknown. To obtain information on this issue, 27 patients with FH from eight unrelated families were analyzed by screening the LDLR (PCR-SSCP and Southern blot) and apoB genes (PCR and restriction enzyme digestion analysis). None of the patients carried either the R3500Q or the R3531C mutation in the apoB gene. By contrast, seven mutations in the LDLR gene were identified, including five missense mutations on exons 4, 6, 8, and 14 (C113R, G266C, A370T, P664L, C690S) and two large deletions (FH Morocco-1 and FH Morocco-2). The two major rearrangements and the missense mutation G266C are novel mutations and could well be causative of FH in the Moroccan population. This study has yielded preliminary information on the mutation spectrum of the LDLR gene among patients with FH in Morocco.
Similar content being viewed by others

Introduction
Familial hypercholesterolemia (FH) is a common autosomal dominant disease associated with elevated low-density lipoprotein (LDL) plasma cholesterol, tendon xanthomatosis, and premature atherosclerosis. The disorder of lipid metabolism in FH is mainly caused by defects in the LDL receptor (LDLR) gene (Goldstein et al. 1995), although it may also be due to alterations in the gene encoding apolipoprotein B (familial defective apoB or FDB) (Soria et al. 1989) or in other unidentified genes (Haddad et al. 1999; Norman et al. 1999; Varret et al. 1999; Hunt et al. 2000).
To date, more than 600 mutations have been described in the LDLR gene (Varret et al. 1998; Heath et al. 2001; databases are accessible at http//www.umd.necker.fr and http//www.ucl.ac.uk/fh). For the apoB gene, only four mutations have been shown to cause FDB: the most frequent is R3500Q (Myant 1993; Leren et al. 1997) and the very rare are R3500W (Gaffney et al. 1995), R3531C (Pullinger et al. 1995), and E3405Q (Fisher et al. 1999).
In this study, we describe the results obtained in a group of 27 Moroccan patients with a clinical diagnosis of FH, and report for the first time the molecular changes in the LDLR gene likely to cause FH in Moroccans. For the purposes of this study, the LDLR gene was screened by PCR-SSCP analysis, DNA sequencing, and Southern blot analysis. In addition, the apoB gene was screened for the presence of the R3500Q and R3531C mutations by PCR and restriction enzyme digestion analysis.
Materials and methods
Patients
All patients gave their informed consent prior to inclusion in the study. Eight Moroccan probands with clinical and biochemical features of FH from eight unrelated families were recruited from the Departments of Endocrinology and Metabolic Disorders at the University Hospitals of Casablanca and Rabat. The diagnostic criteria for FH probands were a high total serum cholesterol (>290 mg/dl), a family history of hypercholesterolemia (at least two affected first-degree relatives), and premature coronary disease, with the presence of arcus cornealis, xanthomas, or xanthelasmas. When members of these families were available, we performed a clinical and biochemical evaluation of FH in relatives and a co-segregation study of the disease and the identified mutation. In total, the study sample comprised 27 FH patients, classified as 20 heterozygotes and seven homozygotes on the basis of clinical and biochemical data (Table 1).
Lipid measurements
Venous blood samples from fasting patients were drawn and serum was isolated after centrifugation at 4°C for 15 min; serum total cholesterol (TC), triglycerides (TG), and high-density lipoprotein cholesterol (C-HDL) were then measured. TC and TG were analyzed by enzymatic methods (Boehringer Mannheim FRG). C-HDL was enzymatically determined by separating HDL from the plasma by precipitation of the (LDL + VLDL) fraction with a phosphotungstic acid/MgCl2 solution (Boehringer Mannheim FRG). Serum LDL cholesterol was calculated according to the Friedewald formula (Friedewald et al. 1972).
DNA analysis
Genomic DNA was isolated from EDTA whole blood samples using a salting-out procedure (Miller et al. 1988).
DNA amplification, SSCP, and DNA sequencing
The promoter and exon 4 were directly sequenced and the rest of the exons of the LDLR gene were screened for the presence of small variations by PCR-SSCP analysis. Amplification of the selected LDLR segments was performed individually by polymerase chain reaction (PCR) using an optimized set of oligonucleotide primers (Hobbs et al. 1992; Jensen et al. 1996) and PCR reaction conditions. For the SSCP analysis, electrophoresis was carried out in a horizontal minigel (Gene Gel Excel 12.5/24 Kit) system (Gene Phor) at 400 V for 15 min and then at 600 V for 1 h at 5–12°C. After the electrophoresis, DNA bands were detected by silver stain. The direct sequencing of the promoter, exon 4, and the fragments showing an altered SSCP pattern was performed in both directions by capillary electrophoresis in an ABI PRISM 310 Genetic Analyzer. Nucleotide positions and variations were numbered as suggested by Yamamoto et al. (1984).
For the apoB gene, the screening for mutations R3500Q and R3531C was performed by PCR-mediated, site-directed mutagenesis and double restriction of a unique PCR product (Rabès et al. 1997).
Southern blot analysis
The LDLR gene was analysed by Southern blot for the presence of large rearrangements. Genomic DNA samples (4 μg) were digested using 5– to 10 U/μg of AvaII, PvuII, EcoRI, and StuI, separated by agarose gel electrophoresis 0.8%, transferred to nylon membranes, and hybridized successively with two 32P-labelled LDLR cDNA probes covering exons 1–10 and exons 10–18.
Confirmation of variations in the LDLR gene
To confirm the existence of variations that create or abolish a given natural restriction site, a rapid PCR-based DNA analysis was performed. Briefly, PCR products of the corresponding exon were digested with the relevant restriction enzymes (according to the manufacturer's instructions), electrophoresed on 2% agarose or 8% polyacrylamide gel, stained with ethidium bromide, and examined under UV illumination.
Results and discussion
Clinical and biochemical features of the probands are shown in Table 1. The mean serum total cholesterol level was 309 and 580 mg/dl for the heterozygous and the homozygous probands, respectively, without lipid-lowering drug therapy.
It is important to note that in our country there is no specialized consultation for dyslipemia patients. Generally, FH patients are seen by cardiologists when cardiovascular complications appear, but the disease's etiology is not always investigated. This could explain the low number of FH patients recruited in our sample.
In this study, our aim was to obtain a preliminary molecular spectrum of the genetic defects causing FH in Morocco. For this purpose, we collected DNA from eight probands belonging to eight unrelated families and from 19 identified FH members of these families. All the probands were screened for R3500Q and R3531C mutations in the apoB gene by PCR-mediated, site-directed mutagenesis. Neither of these FDB-causing mutations were found in our sample. Because we screened the apoB gene only for the R3500Q and R3531C mutations and did not analyse it for other possible mutations, we cannot definitively rule out the presence in our sample of FDB patients with an FH phenotype.
Concerning the analysis of the LDLR gene, we performed the molecular search for mutations in the promoter region and the 18 exons of this gene. Using PCR-SSCP analysis and DNA sequencing, we detected nine variations in the LDLR gene. These included five missense mutations (C113R, G266C, A370T, P664L, C690S) (Table 2) and four silent sequence variations (C6C, L554L, N570N, V632V). Four of the aforementioned missense mutations, C113R, A370T, P664L, and C690S, have been described previously in other populations:
The A370T variation has been shown not to be associated with FH and appears to be a genetic polymorphism in the Caucasian population (Gudnason et al. 1995; Weiss et al. 1998). In our sample, we found this polymorphism in two families in whom it segregated, indicating that this genetic variation is not specific to the Caucasian population.
The P664L mutation has been demonstrated to be causative of FH (Soutar et al. 1989). We found this mutation in one compound heterozygous proband. The proband's brother does not carry this mutation. We have not yet found the other allele defect in these FH patients. At present, we do not know the exact contribution of the P664L mutation to the FH phenotype of the compound heterozygous proband.
The C113R mutation has been described in Dutch and German patients (Heath et al. 2001; Nauck et al. 2001; Fouchier et al. 2001) and C690S in the Dutch population only (Fouchier et al. 2001). It is interesting to note that 300,000 individuals living in the Netherlands are Moroccans, and the finding of these two mutations in our population could be accounted for by the important flux migration to this country. Further investigations must be done to address this issue.
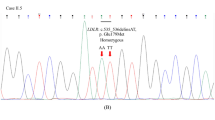
The G266C missense mutation is novel mutation never previously reported, and it has not been found in a sample of 56 healthy Moroccan controls. This mutation is located in repeat VII of the ligand binding domain and changes a conserved glycine to a cysteine. In fact, this domain includes seven repeats and this conserved glycine is found in repeats I, III, V, VI, and VII. This change could interfere with the disulfide bonds pattern, which confers to the receptor an extremely stable structure essential for the protection of the molecule from degradation when it is recycled several times. On the other hand, it has been established that all the repeat sequences from II to VII of the first domain are essential to the optimal binding of apo B in LDL, and replacement of a conserved neutral amino acid (glycine) by an amino acid with a polar residue (cysteine) in repeat VII could also have an effect on apo B binding (Lestavel and Fruchart 1994). The effect of this novel mutation on the functional activity of LDLR is hypothetical. Unfortunately, we were not able to demonstrate the co-segregation of the G266C mutation with the FH phenotype, because family relatives of the proband were not available for genetic testing.
The frequent four silent polymorphisms C6C, L554L, N570N, and V632V were also found in our sample. These polymorphisms were found in more than one individual from different families: C6C was found in one family, N570N in five families, and L554L and V632V in two families.
Southern blot analysis allowed the detection of two different novel partial deletions in two probands, 8M and 12M. Since these deletions have never been reported previously, we have designated these mutations FH Morocco-1 and FH Morocco-2. Southern blotting of the DNA sample from proband 8M showed a heterozygous ~0.9-kb deletion (FH Morocco-1) situated between the 3' end of intron 1 (350 bp downstream from exon 2) and the 5' end of exon 3 (28 bp). The precise limits of this small deletion must be further investigated to assess the potential disease-causing impact of this mutation.
After digestion with EcoRI, the hybridization of the 12M proband's DNA with cDNA probes exon 1–10 showed the loss of the ~9.6-kb fragment and an additional ~3.2-kb fragment. This preliminary screening suggested that 12M was homozygous for a gross deletion (FH Morocco-2), which eliminates exons 2, 3, and 4 and is expected to be the disease-causing mutation.
The molecular status of FH in Morocco has been unknown until now. In this study, we have partially clarified the mutational spectrum of LDLR gene defects in our sample of eight unrelated families, which seems to be heterogeneous. The mutational analysis predicted the potential disease-causing impact of G266C, C113R, and C690S missense mutations in three probands (18M, 6M, 1M) and one partial deletion in one proband (8M). Prediction of the impact of the other rearrangement, found in 12M (deletion of ~0.9 kb), is not possible until the exonic involvement is known. In proband 10M, we cannot attribute the FH phenotype only to the P664L mutation until we have identified the other allele defects present in this family. Therefore, molecular identification of mutant alleles was possible in 75% of all cases. For the remaining cases, the molecular basis of FH remains unknown. This can be explained by the SSCP technique limits to detect all single base changes (Humphries et al. 1997), or by the intervention of other genes causing an FH-like phenotype.
Before any conclusions can be drawn about the molecular status of FH in Morocco, a larger number of clinically diagnosed FH patients will have to be tested.
References
Fisher E, Scharnagl H, Hoffmann MM, et al. (1999) Mutations in the apolipoprotein (apo) B-100 receptor-binding region: detection of apo B-100 (Arg3500→Trp) associated with two new haplotype and evidence that apo B-100 (Glu3405→Gln) diminishes receptor-mediated uptake of LDL. Clin Chem 45:1026–1038
Fouchier SW, Defesche JC, Umans-Eckenhausen MA, Kastelein JJ (2001) The molecular basis of hypercholesterolemia in the Netherlands. Hum Genet 109:602–615
Friedewald WT, Levy FI, Fredrickson DS (1972) Estimation of the concentration of the low density lipoprotein cholesterol in plasma without use of the preparative ultracentrifuge. Clin Chem 18:499–509
Gaffney D, Reid JM, Cameron IM, Vass K, Caslake M, Sheperd J, Packard CJ (1995) Independent mutations at codon 3500 of the apolipoprotein B gene are associated with hyperlipidaemia. Arterioscler Thromb Vasc 15:1025–1029
Goldstein JL, Hobbs HH, Brown MS (1995) Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease, 7th edn. McGraw-Hill, New York, pp 1981–2030
Gudnason V, Patel D, Sun X-M, Humphries S, Soutar AK, Knight BL (1995) Effect of the StuI polymorphism in the LDL receptor gene (Ala370 to Thr) on lipid levels in healthy individuals. Clin Genet 47:68–74
Haddad L, Day IN, Hunt S, Williams RR, Humphries SE, Hopkins PN (1999) LDLR, non-APOB kindred. J Lipid Res 40:1113–1122
Heath KE, Gahan M, Whittal RA, Humphries SE (2001) Low-density lipoprotein receptor gene (LDLR) world-wide web site in familial hypercholesterolemia: update, new features and mutation analysis. Atherosclerosis 154:243–246
Hobbs H, Brown MS, Goldstein JL (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1:4445–4466
Humphries SE, Gudnason V, Whittall R (1997) Single-strand conformation polymorphism analysis with high throughput modifications, and its use in mutation detection in familial hypercholesterolemia. Clin Chem 43:427–435
Hunt SC, Hopkins PN, Bulka K, McDermott MT, Thorne TL, Wardell BB, Bowen BR, Ballinger DG, Skolnick MH, Samuels ME (2000) Genetic localization to chromosome 1p32 of the third locus for familial hypercholesterolemia in Utah kindred. Arterioscler Thromb Vasc Biol 20:1089–1093
Jensen HK, Jensen LG, Hansen PS, Faergeman O, Gregersen N (1996) High sensitivity of the single-strand conformation polymorphism method for detection of sequence variations in the LDL receptor gene validated by DNA sequence. Clin Chem 42:1140–1146
Leren TP, Tonstad S, Gudersen KE, Bakken KS, Rodningen OK, Sundvold H, Ose L, Berg K (1997) Molecular genetics of familial hypercholesterolemia in Norway. J Intern Med 241:185–194
Lestavel S, Fruchart JC (1994) Lipoprotein receptors. Cell Mol Biol 40:461–481
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from nucleated cells. Nucleic Acids Res 16:1215
Myant NB (1993) Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolemia. Atherosclerosis 104:1–18
Nauck MS, Köster W, Dörfer K, Eckes J, Scharnagl H, Gierens H, Nissen H, Nauck MA, Wieland H, März W (2001) Identification of recurrent and novel mutations in the LDL receptor gene in German patients with familial hypercholesterolemia. Hum Mutat 18:165–166
Norman D, Sun XM, Bourbon M, Knight BL, Naoumova RP, Soutar AK (1999) Characterization of a novel cellular defect in patients with phenotypic homozygous familial hypercholesterolemia. J Clin Invest 104:619–628
Pullinger CR, Hennessy LK, Chatterton JE, Liu W, Love JA, Mendel CM, Frost PH, Malloy MJ, Schumaker VN, Kane JP (1995) FDB: identification of a new mutation that decrease LDL-receptor binding affinity. J Clin Invest 95:1225–1234
Rabès JP, Varret M, Saint-Jore B, Erlich D, Jondeau G, Giraudet P, Junien C, Boileau C (1997) Familial ligand-defective apolipoprotein B-100: simultaneous detection of the Arg3500→Gln and Arg3531→Cys mutations in a French population. Hum Mut 10:160–163
Soria LF, Ludwig EH, Clarke HRG, Vega GL, Grundy SM, McCarty BJ (1989) Association between a specific apo B mutation and familial defective apoB100. Proc Natl Acad Sci U S A 86:587–591
Soutar AK, Knight BL, Patel DD (1989) Identification of a point mutation in growth factor receptor C of the low density lipoprotein-receptor gene in a patient with homozygous familial hypercholesterolemia that affects ligand binding and intracellular movement of receptors. Proc Natl Acad Sci U S A 86:4166–4170
Varret M, Rabès JP, Kotze MJ, Baron H, Cenarro A, Descamps O, Ebhardt M, Nondelijn JC, Kostner GM, Miyake Y, Pocovi M, Shmidt H, Schuster H, Stuhrmann M, Yamamura T, Junien C, Beroud C, Boileau C (1998) LDLR database (second edition): new additions to the database and the software, and results of the first molecular analysis. Nucleic Acids Res 26:248–252
Varret M, Rabès JP, Saint-Jore B, Cenarro A, Marinoni JC, Civeira F, Devillers M, Krempf, Culon M, Thiart R, Kotze MJ, Schmidt H, Buzzi JC, Kostner GM, Bertoleni S, Rosa A, farnier M, Martinez M, Junien C, Boileau C (1999) A third major locus for autosomal dominant hypercholesterolemia maps to 1p34.1–32. Am J Human Gene 64:1378–1387
Weiss N, Binder G, Keller C (1998) Heterozygosity for the missense mutation Ala370→Thr in exon 8 of the low density lipoprotein receptor gene does not cause hypercholesterolemia. Eur J Med Res 3:20–24
Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, Russell DW (1984) The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell 39:27–38
Acknowledgements
We thank the many clinicians who provided the blood samples and clinical data of their FH patients. We also thank Dr. A. Cenarro for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
El Messal, M., Aït Chihab, K., Chater, R. et al. Familial hypercholesterolemia in Morocco: first report of mutations in the LDL receptor gene. J Hum Genet 48, 199–203 (2003). https://doi.org/10.1007/s10038-003-0010-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0010-x
Keywords
This article is cited by
-
Common and rare single nucleotide polymorphisms in the LDLR gene are present in a black South African population and associate with low-density lipoprotein cholesterol levels
Journal of Human Genetics (2014)
-
Familial hypercholesterolemia associated with severe hypoalphalipoproteinemia in a Moroccan family
Journal of Genetics (2007)
-
ApoB-100 R3500Q mutation in the Lebanese population: Prevalence and historical review of the literature
Molecular Biology Reports (2007)
