
Abstract
This manuscript describes the preparation of an advanced intermediate toward the total synthesis of citrinadin A, featuring a [3+2] cycloaddition employing in situ generation of the dipole.
Similar content being viewed by others
Introduction
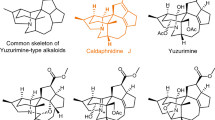
In the early 2000’s, Kobayashi and coworkers1 reported the isolation of two secondary metabolites, citrinadin A and B, from the marine fungus Penicillium citrinum N059. Structural determination efforts by Kobayashi revealed that the two congeners possess the same complex spirooxindole-containing pentacyclic core and differ only by an N,N-dimethylvaline appendage resident on citrinadin A (cf. 1 and 2, Figure 1, top).

The citrinadins.
The interesting structural features of these compounds have led several groups to begin exploring their total syntheses. Recently our efforts and those of the Martin group led to completed syntheses of citrinadin B and A, respectively. These efforts independently led to the conclusion that the structures assigned by Kobayashi were sound in terms of connectivity, but required revision at the stereochemical level.2, 3, 4, 5 As illustrated in Figure 1 (bottom), the reassigned structures of citrinadins A and B differ at the stereogenic centers residing within the pentacyclic core. Herein, we report synthetic efforts toward citrinadin A (3) that have now resulted in the stereoselective preparation of its core structure.
Results and Discussion
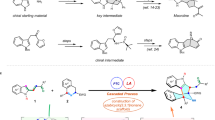
Citrinadin A possesses several synthetically challenging features, including an epoxy ketone side chain and a rare N,N-dimethylvaline ester. We envisioned that both of these moieties would be installed at a late stage, a maneuver that was particularly important for the delicate epoxy ketone. Thus, from a retrosynthetic perspective, citrinadin A was seen as arising from 5, wherein an aryl bromide is poised for installation of the epoxy ketone side chain, and a masked alcohol will enable installation of the ester through a Mitsunobu reaction (Scheme 1). Epoxide 5 would be available from pentacycle 6 via reductive cleavage of the isoxazolidine N–O bond and subsequent quinolizidine formation. The isoxazolidine 6 would result from the intermolecular [3+2] cycloaddition of enone 7 and nitrone 8. The requisite enone is the same as that employed in our previous synthesis of citrinadin B, whereas the nitrone was expected to arise from known diol 11 (vide infra).
During our initial studies towards citrinadin A,6 we attempted to preform the nitrone before cycloaddition. Although this approach was successful in our synthesis of citrinadin B, under similar conditions 8 performed poorly. After considerable experimentation, we eventually turned to a strategy that employed in situ generation of the requisite nitrone.6 As illustrated retrosynthetically in Scheme 2, delivery of the nitrone would follow from desilylative cyclization of tert-butyl dimethylsilyl (TBS)-protected oxime 10, which in turn would arise from the known diol 11.7
In the forward sense, known diol 11 was converted to the known lactone 12 using a sequence previously reported by Nakamura et al.7, which involved exposure to pyridinium p-toluenesulfonate (PPTS) in refluxing dichloroethane, followed by tert-butyl diphenylsilyl (TBDPS) protection (Scheme 3) (nitrone precursor 10 was prepared using racemic ethyl-3-hydroxybutyrate in lieu of (S)-3-hydroxybutanoate as described in Nakamura et al.7). Subsequent treatment of 12 with diisobutyl aluminum hydride (DIBAL-H) furnished the corresponding lactol 13 as a 1:1 mixture of diastereomers which, upon exposure to hydroxylamine hydrochloride, underwent smooth conversion to oxime 14 as a 1:1 mixture of E and Z isomers. Conversion of 14 into a substrate suitable for desilylative cyclization required initial TBS protection of the oxime alcohol and activation of the remaining secondary alcohol through tosylation.
Having prepared racemic nitrone precursor 10, we began to explore its ability to undergo cycloaddition with the previously prepared racemic enone 7. As alluded to above, attempts to employ the preformed nitrone met with limited success. Thus, we began investigating generation of the ntirone in situ and, after an exhaustive screen of fluorine sources, solvents, and ratios of 7–10, we discovered that tetrabutylammonium difluorotriphenylsilicate (TBAT) in benzene with three equivalents of 10 gave the best overall result (Scheme 4).6
We were pleased to learn that unlike the nitrone cycloaddition used en route to citrinadin B, which required 9 days, the reaction of 7 with 10 furnished the desired cycloadduct in just 24 h.8 Although 15b was the minor diastereomer, we have demonstrated in preliminary studies that use of enantioenriched 7,5 gives a diastereomeric ratio (d.r.) of 3:2, whereas use of enantioenriched 7 and enantioenriched 10, derived from commercially available chiral ethyl-3-hydroxybutyrate, gives a d.r. of 2:1. Fortunately, 15a and 15b were separable via flash column chromatography, thus in these early studies, we were able to continue advancing the desired cycloadduct (15b) towards citrinadin A.
Toward this end, 15b was treated with trimethylsulfoxonium iodide and sodium hydride (Scheme 5). The derived Corey–Chaykovsky adduct (16) was stereoselectively produced in good yield.9, 10 Subsequent exposure of 16 to trimethylsilyl chloride and NaI promoted intramolecular attack on the epoxide by the isoxazolidine nitrogen to furnish an ammonium salt (17), which was then reduced with zinc in AcOH to give diol 18, thereby completing construction of the core ring system.11 This began setting the stage for eventual incorporation of the angular nitrogen atom. Thus, the least hindered alcohol in 18 was activated for displacement by treating with MsCl and Et3N. Subsequent exposure of the derived mesylate to K2CO3 promoted the formation of key intermediate 19, wherein the ring-fusion epoxide stands ready to mediate incorporation of the final nitrogen.
Conclusion
In summary, we have prepared a late-stage intermediate en route to citrinadin A with an approach that employs an intermolecular [3+2] nitrone cycloaddition. The latter was found to best proceed under conditions wherein the nitrone is produced in situ and, although the cycloaddition was performed with racemic substrates, its stereochemical outcome clearly indicates that enantioenriched substrates will selectively deliver material required for preparation of the natural product. These efforts have also illustrated that the derived cycloadduct can be advanced to an intermediate (19) that contains all of the functional groups required for conversion to citrinadin A. The aryl bromide will allow for attachment of the epoxy ketone, removal of the TBDPS group will set the stage for introduction of the valine ester via a Mitsunobu reaction, and opening of the ring-fusion epoxide will provide access to the requisite anti relationship between the methylamine and alcohol substituents. Work toward these ends is underway and will be reported in due course.
Experimental Procedure
General
Unless otherwise noted, all reactions have been carried out with distilled and degassed solvents under an atmosphere of dry N2 in oven- (135 °C) or flame-dried glassware with standard vacuum-line techniques. Triethylamine, diisopropylamine and methanol were dried over calcium hydride and freshly distilled. Benzene, tetrahydrofuran, methylene chloride, toluene, acetonitrile and diethyl ether were dried using a solvent purification system manufactured by SG Water U.S.A., LLC (Nashua, NH, USA) as follows: tetrahydrofuran, diethyl ether, acetonitrile and methylene chloride were passed through two packed columns of neutral alumina, whereas benzene and toluene were passed through a column of alumina and a column of Q5. All other commercially available reagents were used as received.
Unless otherwise stated, all reactions were monitored by TLC using glass-backed extra hard layer, 60 Å plates (Indicator F-254, 250 μm, Silicycle, Ville de Québec, QC, Canada). Column or flash chromatography was performed with the indicated solvents using Silicycle SiliaFlash P60 (230–400 mesh) silica gel as the stationary phase. All melting points were obtained on a Gallenkamp capillary melting point apparatus (model: MPD350.BM2.1, Sanyo Electric Co., Ltd., Osaka, Japan) and are uncorrected. IR spectra were obtained using a Nicolet Avatar 320 FTIR (Thermo Electron Corporation, Madison, WI, USA) or Bruker Tensor 27 FTIR (Bruker Optics Inc., Billerica, MA, USA). 1H and 13C NMR spectra were recorded on a Varian Inova 500, Varian Inova 400, Varian Inova 400 autosampler, or Varian Inova 300 spectrometer (Varian, Inc., Palo Alto, CA, USA). Chemical shifts (δ) are reported in p.p.m. relative to internal residual solvent peaks from indicated deuterated solvents. Coupling constants (J) are reported in Hertz (Hz) and are rounded to the nearest 0.1 Hz. Multiplicities are defined as: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, dd=doublet of doublets, dt=doublet of triplets, ddd=doublet of doublet of doublets, dddd=doublet of doublet of doublet of doublets, br=broad, app=apparent, par=partial. HRMS were performed at the Central Instrument Facility by Donald L. Dick of Colorado State University.
Lactol 13
Lactone 12 (3.2 g, 8.7 mmol) in dichloromethane (90 ml) was cooled to −78 °C for the addition of diisobutylaluminum hydride solution (1 M in hexanes, 9.6 ml, 9.6 mmol). This was stirred cold for 2 h at which time the reaction was quenched at −78 °C with methanol (9 ml) and allowed to warm to room temperature. The resulting reaction solution was vigorously stirred with an added solution of Rochelle’s salt (9 g in 100 ml H2O) for 1 h. Following separation of the layers, the aqueous portion was extracted with diethyl ether, and the combined organics were washed with brine, dried over sodium sulfate (Na2SO4) and concentrated to provide lactol 13 as a 1:1 mixture of anomers (3.2 g, 100% yield), which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 7.71–7.68 (m, 8H), 7.46–7.36 (m, 12H), 5.30 (br, t, 1H), 4.50 (ddd, J=9.7, 6.6, 2.1 Hz, 1H), 4.22 (m, 1H), 3.91 (m, 2H), 3.79 (tt, J=10.9, 4.8 Hz, 1H), 3.31 (dqd, J=11.6, 5.9, 1.9 Hz, 1H), 3.01 (t, J=2.6 Hz, 1H), 2.09 (ddt, J=12.2, 4.4, 2.1 Hz, 1H), 1.98 (ddt, J=12.8, 4.7, 1.7 Hz, 1H), 1.76 (m, 2H), 1.60 (dddd, J=12.9, 11.0, 3.6, 2.0 Hz, 1H), 1.48~1.26 (m, 3H), 1.19 (d, J=6.2 Hz, 3H), 1.11 (d, J=6.3 Hz, 3H), 1.08 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.83, 134.60, 134.48, 134.24, 134.13, 129.84, 129.82, 129.70, 129.69, 127.76, 127.74, 127.67, 127.65, 94.39, 92.93, 68.28, 68.16, 65.14, 64.16, 43.30, 42.61, 42.29, 39.51, 27.07, 27.01, 21.51, 21.27, 19.24, 19.19; IR (thin film): 3398 (br, m), 2932 (m), 2858 (m), 1428 (m), 1112 (s), 702 (s) cm−1; HRMS (ESI) Calcd. for C22H30NaO3Si [M+Na]: 393.1858. Found: 393.1862.
Oxime 14
The lactol 13 (2.8 g, 7.6 mmol) was then taken up in pyridine (15 ml). To this was added anhydrous magnesium sulfate (MgSO4) (1.8 g, 15.2 mmol) and hydroxylamine hydrochloride (791 mg, 11.4 mmol). This was allowed to stir at room temperature for 36 h. To work up, the reaction mixture was filtered through Celite, which was subsequently washed with 1:1 dichloromethane/hexanes and then dichloromethane. The resulting solution was concentrated, azeotroped with hexanes, and subjected to column chromatography (5%→10%→15%→30%→50% ethyl acetate/hexanes) to provide oxime 14 as an oil in a 1:1 mixture of E/Z isomers (2.9 g, 100%). 1H NMR (400MHz, CDCl3) δ 9.16 (br, s, 1H), 8.81 (br, s, 1H), 7.71~7.66 (m, 8H), 7.45~7.35 (m, 12H), 7.26 (t, J=6.3 Hz, 1H), 6.67 (t, J=5.5 Hz, 1H), 4.15 (m, 2H), 3.94 (m, 2H), 2.63~2.41 (m, 5H), 2.30 (ddd, J=14.6 Hz, 6.6 Hz, 4.4 Hz, 1H), 1.66–1.52 (m, 4H), 1.069 (s, 9H), 1.065 (s, 9H), 1.05 (d, J=6.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 149.54, 148.81, 148.64, 136.02, 135.99, 133.48, 133.47, 133.38, 133.33, 130.09, 127.89, 69.73, 69.24, 64.60, 64.52, 45.08, 44.63, 36.70, 32.38, 27.11, 23.78, 19.35; IR (thin film): 3268 (br, m), 2931 (m), 2858 (m), 1428 (m), 1112 (s), 738 (m), 703 (s) cm−1; HRMS (ESI) Calcd. for C22H31NNaO3Si [M+Na]: 408.1969. Found: 408.1971.
Nitrone precursor 10
To a solution of oxime 14 (2.9 g, 7.6 mmol, azeotroped in toluene) in dichloromethane (75 ml) was added imidazole (1 g, 15.2 mmol). The solution was cooled to 0 °C in an ice bath and tertbutyldimethylsilyl chloride (0.6 g, 4 mmol) was added. The reaction was stirred cold for 45 min. Then, another portion of tertbutyldimethylsilyl chloride (0.6 g, 4 mmol) was added and the reaction was allowed to gradually come to room temperature overnight. The reaction was quenched with ammonium chloride (NH4Cl) (50% sat., aq., soln.) and the aqueous layer was extracted with diethyl ether. The organic extracts were washed with brine, dried (Na2SO4), concentrated and purified by flash chromatography (0%→2%→4%→8%→15% ethyl acetate/hexanes) to yield the TBS-protected oxime as an oil with a 1:1 mixture of olefin isomers (3 g, 79% yield). 1H NMR (400 MHz, CDCl3) δ 7.73~7.68 (m, 8H), 7.47~7.37 (m, 12H), 7.33 (t, J=6.3 Hz, 1H), 6.85 (t, J=5.4 Hz, 1H), 4.17 (m, 2H), 4.01 (m, 2H), 2.70 (ddd, J=15.6 Hz, 7.1 Hz, 5.5 Hz, 1H), 2.57 (ddd, J=15.7 Hz, 9.9 Hz, 5.1 Hz, 1H), 2.53 (dt, J=14.5 Hz, 6.6 Hz, 1H), 2.5 (d, J=3.0 Hz, 1H), 2.31 (ddd, J=14.5 Hz, 5.9 Hz, 4.3 Hz, 1H), 2.27 (d, J=3.2 Hz, 1H), 1.61 (m, 5H), 1.093 (d, J=6.0 Hz, 3H), 1.09 (s, 18H), 1.07 (d, J=6.4, 3H), 0.92 (s, 9H), 0.91 (s, 9H), 0.14 (s, 6H), 0.13 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 152.31, 152.10, 136.06, 136.01, 133.53, 133.46, 133.38, 133.25, 130.13, 130.11, 127.93, 127.90, 69.99, 69.48, 64.49, 64.31, 44.99, 44.03, 36.52, 32.86, 27.14, 26.23, 26.16, 23.82, 19.39, 19.34, 18.29, 18.23, −5.14, −5.16; IR (thin film): 3452 (br, m), 2931 (s), 2858 (s), 1472 (m), 1112 (s), 703 (s) cm−1; HRMS (ESI) Calcd. for C28H46NO3Si2 [M+H]: 500.3020. Found: 500.3016.
A solution of the TBS-protected oxime (1.1 g, 3.6 mmol) and pyridine (10 ml) in dichloromethane (30 ml) was cooled to 0 °C in an ice bath. To this was added tosyl chloride (2.7 g, 14.4 mmol). The solution was allowed to gradually warm to room temperature overnight (20 h). The reaction was quenched with CuSO4 (10% aq. soln.) and the aqueous layer was extracted with diethyl ether. The combined organics were washed with water and brine, dried (Na2SO4) and concentrated. The crude oil was purified by flash chromatography (1%→2%→3% ethyl acetate/hexanes) to provide tosylate 10 (1.3 g, 81% yield) as a 1:1 mixture of E/Z olefin isomers, which was either used immediately or temporarily stored as a solution (1 M in benzene, over molecular sieves) in the freezer. 1H NMR (400 MHz, CDCl3) δ 7.55~7.49 (m, 12H), 7.34~7.21 (m, 12H), 7.16 (t, J=6.2 Hz, 1H), 7.13 (d, J=7.4 Hz, 4H), 6.70 (t, J=5.2 Hz, 1H), 4.46 (m, 2H), 3.75 (quintet, J=5.7 Hz, 2H), 2.30 (dt, J=16.0 Hz, 5.4 Hz, 1H), 2.29 (s, 6H), 2.26 (dt, J=16.0 Hz, 5.6 Hz, 1H), 2.13 (dt, J=14.6 Hz, 6.3 Hz, 1H), 2.06 (ddd, J=14.6 Hz, 6.2 Hz, 4.9 Hz, 1H), 1.74 (ddd, J=14.5 Hz, 7.5 Hz, 6.1 Hz, 1H), 1.68 (dd, J=14.0 Hz, 7.2 Hz, 1H), 1.49 (tt, J=14.0 Hz, 5.7 Hz, 2H), 0.93 (d, J=6.3 Hz, 3H), 0.907 (s, 9H), 0.903 (s, 9H), 0.89 (d, J=3.0 Hz, 3H), 0.79 (s, 9H), 0.77 (s, 9H), 0.005 (d, J=2.5 Hz, 6H), 0.00 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 18.20, 18.25, 19.35, 19.39, 21.27, 21.47, 21.74, 26.15, 26.22, 27.08, 33.09, 37.22, 44.25, 44.62, 68.30, 68.71, 77.88, 77.96, 127.70, 127.72, 127.81, 127.83, 127.85, 129.81, 129.93, 129.97, 133.64, 133.70, 133.77, 134.81, 134.87, 135.93, 135.95, 135.98, 144.46, 144.49, 152.09, 152.18, −5.11, −5.15; IR (thin film): 2931 (m), 2858 (m), 1363 (m), 1177 (s), 1111 (m), 919 (m), 703 (m) cm−1; HRMS (ESI) Calcd. for C35H52NO5SSi2 [M+H]: 654.3105. Found: 654.3113.
Isoxazolidines 15a and 15b
To a solution of the nitrone precursor 10 (118 mg, 0.18 mmol) in benzene (900 μl) was added MgSO4 (43 mg, 0.36 mmol) and the enone 7 (24.5 mg, 0.06 mmol). Lastly, tetrabutylammonium difluorotriphenylsilicate (97 mg, 0.18 mmol) was added in two portions. This stirred at ambient temperature overnight (17 h). At that time, the heterogeneous mixture was diluted with dichloromethane (1 ml) and filtered through a celite plug. The filtrate was directly adsorbed onto silica gel and purified by flash chromatography (0%→2.5%→5%→→20% ethyl acetate/hexanes) to provide two isoxazolidine diastereomers (39 mg total, 84% yield, 15a:15b=d.r. 7:3), the desired isoxazolidine 15b (12 mg) and the undesired diastereomer 15a (27 mg), both as white foams. Isoxazolidine 15a: 1H NMR (400 MHz; CDCl3) δ 7.70 (m, J=1.5 Hz, 4H), 7.57 (dd, J=7.6, 0.9 Hz, 1H), 7.44–7.27 (m, 10H), 7.23 (d, J=7.2 Hz, 2H), 6.91 (t, J=7.9 Hz, 1H), 5.42 (m, 2H), 3.98 (br s, 1H), 3.79 (br m, 1H), 3.42 (br m, 2H), 2.90 (s, 2H), 2.44 (br m, 1H), 2.02 (d, J=14.4 Hz, 1H), 1.91 (d, J=14.4 Hz, 1H), 1.55 (m, 1H), 1.25 (m, 1H), 1.13 (m, 12H), 1.01 (s, 3H), 0.97 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 14.35, 19.24, 19.99, 20.62, 22.44, 27.10, 32.31, 38.16, 39.59, 43.70, 44.96, 48.24, 48.67, 55.01, 61.28, 65.67, 77.36, 93.73, 102.10, 123.49, 125.97, 126.57, 127.20, 127.78, 128.65, 129.80, 129.86, 133.98, 134.02, 134.40, 136.01, 136.06, 137.78, 141.04, 180.54, 213.66; IR (thin film): 2930 (s), 1751 (s), 1718 (s), 1450 (m), 1104 (s), 733 (s) cm−1; HRMS (ESI) Calcd. for C44H49BrN2O4Si [M+H+2]: 779.2723. Found: 779.2707. Isoxazolidine 15b: 1H NMR (400 MHz; CDCl3) δ 7.72–7.67 (m, 4H), 7.46–7.39 (m, 6H), 7.37 (dd, J=8.2, 0.9 Hz, 1H), 7.33–7.29 (m, 2H), 7.26–7.22 (m, 3H), 6.82 (d, J=7.4 Hz, 1H), 6.71 (t, J=7.8 Hz, 1H), 5.40 (d, J=3.4 Hz, 2H, 4.03 (br m, 1H), 3.93 (br m, 1H), 3.37 (m, 2H), 3.29 (d, J=18.9 Hz, 1H), 2.53 (d, J=18.9 Hz, 1H), 2.26 (br m, 1H), 1.88 (br s, 2H), 1.68 (d, J=14.2 Hz, 1H), 1.31 (t, J=12.7 Hz, 1H), 1.23 (s, 3H), 1.19 (d, J=5.7 Hz, 3H), 1.14 (s, 9H), 0.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 216.00, 178.69, 141.09, 137.59, 135.97, 135.92, 135.19, 134.59, 134.00, 133.80, 130.02, 129.90, 128.64, 127.88, 127.82, 127.28, 126.84, 123.46, 122.43, 102.48, 93.00, 66.02, 60.98, 54.39, 49.61, 49.56, 45.10, 43.08, 42.08, 39.62, 32.07, 27.08, 20.84, 19.93, 19.76, 19.43; IR (thin film): 3278 (br, w), 2963 (m), 1748 (s), 1722 (s), 1450 (m), 1125 (s), 1068 (s), 729 (s) cm−1; HRMS (ESI) Calcd. for C44H49BrN2O4Si [M+H+2]: 779.2723. Found: 779.2703.
Spiroepoxide 16
To a solution of trimethylsulfoxonium iodide (145.3 mg, 0.66mmol) in dimethylsulfoxide (2.2 ml) was added NaH (60% in mineral oil, 26.4 mg, 0.66 mmol). After stirring at room temperature for 4 h, the resulting homogeneous solution was added a solution of the ketone 15b (170 mg, 0.22 mmol) in tetrahydrofuran (4.4 ml) and cooled to 0 °C in an ice bath to produce an opaque white solution. This was stirred at ambient temperature for 36 h, cooled to 0 °C and quenched with saturated NaHCO3 solution. The mixture was diluted with H2O and the aqueous layer was extracted with ethyl acetate. The combined organic portions were washed with brine solution, dried (Na2SO4), concentrated in vacuo and purified by flash chromatography (4%→8%→12%→30% ethyl acetate/hexanes) to provide the desired product 16 as a white foam (140 mg, 80% yield). 1H NMR (400 MHz; CDCl3) δ 7.72 (m, 4H), 7.42 (m, 6H), 7.34 (d, J=8.3 Hz, 1H), 7.30 (m, 3H), 7.24 (d, J=7.1 Hz, 1H), 7.21 (m, 2H), 6.87 (t, J=7.8 Hz, 1H), 5.35 (s, 2H), 3.96 (s, 1H), 3.34~3.24 (m, 4H) 3.05 (s, 1H), 2.55 (d, J=14.6 Hz, 1H), 2.43 (br s, 1H), 2.20 (d, J=14.6 Hz, 1H), 1.90 (m, 1H), 1.84 (m, 1H), 1.62 (m, 1H), 1.28~1.09 (m: 9H;3H;3H;1H), 0.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 180.69, 141.14, 137.82, 136.56, 136.00, 134.11, 134.00, 129.87, 128.60, 127.78, 127.14, 126.68, 124.07, 122.57, 102.19, 91.85, 77.36, 65.72, 65.57, 59.19, 56.05, 55.21, 50.44, 49.64, 44.88, 39.55, 38.26, 36.92, 36.21, 34.81, 34.66, 32.45, 31.72, 29.20, 27.12, 27.06, 25.42, 23.48, 22.79, 21.00, 20.85, 19.32, 18.91, 14.26, 11.57; IR (thin film): 2930 (s), 1723 (s), 1450 (s), 1105 (s), 734 (s), 703 (s) cm−1; HRMS (ESI) Calcd. for C45H52BrN2O4Si [M+H+2]: 793.2880. Found: 793.2870.
Ammonium Salt 17
To a solution of the epoxide 16 (30 mg, 0.04 mmol) in acetonitrile/tetrahydrofuran (3:1, 0.6 ml) cooled to 0 °C in an ice bath was added NaI (30 mg, 0.2 mmol) and TMSCl (12.5 ml, 0.1 mmol). The clear solution became yellow and a precipitate formed. After stirring for 4 hours, the reaction was diluted with Et2O and washed with saturated Na2S2O3 solution. (Note: DO NOT wash with brine solution.) The aqueous layer was extracted with ethyl acetate. The combined organic extracts were concentrated in vacuo and purified by flash chromatography (20%→40%→60%→ 80%→100% ethyl acetate/hexanes→5% methanol/dichloromethane) to provide the desired product 17 as a clear glass (26 mg, 70% yield). 1H NMR (400 MHz; CDCl3) δ 7.66 (m, J=1.6 Hz, 4H), 7.54 (dd, J=7.6, 1.1 Hz, 1H), 7.50–7.40 (m, 6H), 7.31 (dd, J=8.2, 1.1 Hz, 1H), 7.28 (m, 1H), 7.22 (m, 1H), 7.13 (m, 2H), 6.89 (dd, J=8.2, 7.6 Hz, 1H), 5.31 (s, 2H), 5.16 (d, J=11.9 Hz, 1H), 4.10 (d, J=11.9 Hz, 1H), 4.01 (m, 2H), 3.82 (m, 1H), 2.99 (q, J=13.3 Hz, 2H), 2.81 (dd, J=12.4, 8.0 Hz, 1H), 2.49~2.41 (m, 2H), 2.21 (m, 1H), 2.15 (dt, J=15.6, 5.4 Hz, 1H), 1.84 (dd, J=15.1, 4.6 Hz, 1H), 1.24 (d, 3H), 1.07 (s, 9H), 1.04 (s, 3H), 1.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 193.83, 180.27, 140.10, 137.40, 135.83, 134.43, 132.87, 132.76, 130.50, 130.48, 128.71, 128.24, 128.21, 127.40, 127.22, 126.40, 123.95, 108.64, 102.07, 98.63, 86.22, 77.36, 75.58, 68.75, 65.64, 64.58, 61.68, 51.33, 46.51, 45.11, 36.22, 35.10, 29.86, 26.95, 26.35, 26.28, 20.70, 19.15, 13.36; HRMS (ESI) Calcd. for C45H52BrN2O4Si [M+H+2]: 793.2874. Found: 793.2861.
Diol 18
To a solution of this ammonium salt 17 (8 mg, 0.009 mmol) in tetrahydrofuran/acetic acid (180 μl/240 μl) was added activated zinc powder (4 mg, ~0.05 mmol). The reaction mixture was vigorously stirred under nitrogen for 18 h at ambient temperature, after which time the solvent had evaporated. The solvent mixture of tetrahydrofuran/acetic acid (180 μl/240 μl) was added again, and the reaction was stirred in a sealed flask for 2 more days. This was then filtered through a pad of Celite, rinsed with dichloromethane and concentrated in vacuo. The resulting yellowish oil was dissolved in dichloromethane, washed with NaHCO3 (sat. aq. soln.) and the aqueous portion extracted with ethyl acetate/dichloromethane. The combined organic layers were concentrated and purified by flash chromatography (basic Al2O3, 0%→1%→2% methanol/dichloromethane) to provide the desired product 18 as a clear oil (5 mg, 71% yield). 1H NMR (400 MHz; CDCl3) δ 7.65 (m, 4H), 7.48 (dd, J=7.5, 1.1 Hz, 1H), 7.39 (m, 6H), 7.32 (dd, J=8.1, 1.1 Hz, 1H), 7.28 (m, 2H), 7.21 (m, 3H), 6.93 (dd, J=8.1, 7.5 Hz, 1H), 5.36 (s, 2H), 4.90 (d, J=2.7 Hz, 1H), 4.31 (b s, 1H), 3.92 (tt, J=11.0, 4.5 Hz, 1H), 3.23 (d, J=10.2 Hz, 1H), 2.98 (m, 1H), 2.75 (tt, J=11.1, 2.8 Hz, 1H), 2.47 (d, J=14.4 Hz, 1H), 2.22 (d, J=10.2 Hz, 1H), 2.02 (d, J=14.4 Hz, 1H), 1.82 (ddt, J=12.2, 4.5, 2.4 Hz, 1H), 1.74 (td, J=11.7, 4.7 Hz, 1H), 1.63~1.57 (m, 2H), 1.45 (dd, J=13.0, 3.4 Hz, 1H), 1.32 (m, 1H), 1.22 (s, 3H), 1.05 (s, 9H), 0.87 (s, 3H), 0.75 (d, J=6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 185.31, 140.15, 137.31, 136.57, 135.89, 135.84, 134.76, 134.54, 133.70, 129.74, 129.72, 128.66, 127.71, 127.68, 127.27, 126.87, 126.59, 124.01, 102.23, 82.93, 81.97, 77.36, 66.16, 61.05, 56.35, 55.56, 52.49, 46.38, 46.06, 45.22, 43.74, 41.96, 36.13, 29.86, 27.19, 27.14, 21.68, 19.27, 11.79; IR (thin film): 3390 (br), 3069 (w), 2959, 2929, 2856, 1686 (s), 1450, 1359, 1106, 1064, 734, 701 cm−1; HRMS (ESI) Calcd. for C45H53BrN2O4Si [M+H+2]: 795.3036. Found: 795.3028.
Ring-fusion epoxide 19
To a solution of the diol 18 (116 mg, 0.141 mmol) in dichloromethane (3.0 ml) at room temperature was added MsCl (32.5 μl, 0.423 mmol), dropwise, and let stir for 5 min. To the resulting clear yellow solution was added triethylamine (0.2 ml, 1.41 mmol), dropwise, to give a dark orange, clear solution that was allowed to stir for 2 h, after which equal amounts of MsCl and triethylamine were again added and allowed to stir for 3 h. Methanol (3.0 ml) was added, followed by K2CO3 (0.083 g, 0.60 mmol), to give a cloudy orange reaction mixture. After 16 h, the reaction was quenched with NaHCO3 (sat. aq. soln., 10 ml), extracted with dichloromethane/ethyl acetate, with combined organics washed with brine, dried over Na2SO4 and concentrated in vacuo. The resulting residue was dry loaded on silica and purified via flash column chromatography (5→10→20→30→50% ethyl acetate/hexanes+1% triethylamine ea.) to furnish the desired epoxide as on off-white foam (83.0 mg, 76% yield) and recovered starting material (29 mg). Note: To ensure reproducible 1H NMR data, product was taken up in 10% methanol/dichloromethane, passed through a plug of basic alumina with 10% methanol/dichloromethane as eluent and concentrated in vacuo. 1H NMR (500 MHz, CDCl3) δ 7.67 (ddd, J=8.1, 4.2, 1.5 Hz, 4H), 7.45~7.40 (m, 2H), 7.40~7.35 (m, 4H), 7.31 (dd, J=8.2, 1.1 Hz, 1H), 7.25 (d, J=6.6 Hz, 1H), 7.23~7.14 (m, 5H), 7.08 (dd, J=7.6, 1.2 Hz, 1H), 6.84 (t, J=7.8 Hz, 1H), 5.31 (s, 2H), 3.90 (tt, J=10.4, 4.4 Hz, 1H), 3.16~3.06 (m, 2H), 3.02 (dq, J=7.3, 3.9, 3.5 Hz, 1H), 2.60 (d, J=14.6 Hz, 1H), 2.58~2.52 (m, 1H), 2.19 (d, J=14.6 Hz, 1H), 1.92~1.77 (m, 3H), 1.69 (ddd, J=12.2, 10.5, 4.5 Hz, 1H), 1.64~1.57 (m, 1H), 1.33 (dt, J=12.7, 10.6 Hz, 1H), 1.06 (s, 9H), 1.01 (s, 3H), 0.94 (s, 3H), 0.71 (d, J=6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 179.47, 140.70, 138.06, 137.99, 135.87, 135.82, 134.61, 134.46, 134.41, 134.12, 129.76, 129.73, 129.16, 128.41, 128.38, 128.35, 127.71, 127.68, 127.66, 127.07, 126.98, 125.43, 125.27, 122.51, 101.86, 70.14, 65.93, 65.55, 59.07, 52.47, 49.54, 47.93, 44.97, 44.66, 43.10, 42.15, 40.82, 31.89, 31.66, 27.11, 25.95, 21.60, 20.34, 19.23, 11.44. IR (thin film): 3069 (w), 2931, 2856, 2247 (w), 1724, 1461, 1333, 1109 (s), 1070, 732, 702, 511 cm−1; HRMS (ESI) Calcd. for C45H52BrN2O3Si [M+H+2]: 777.2931. Found: 777.2910.

Retrosynthetic analysis of citrinadin A.

Retrosynthesis of nitrone 9.

Synthesis of the nitrone precursor.

[3+2] Nitrone cycloaddition.

Completion of the core ring system.
References
Tsuda, M. et al. A novel pentacyclic alkaloid from marine-derived fungus Penicillium citrinum. Org. Lett. 6, 3087–3089 (2004).
Bian, Z., Marvin, C. C. & Martin, S. F. Enantioselective total synthesis of (–)-citrinadin A and revision of its stereochemical structure. J. Am. Chem. Soc. 135, 10886–10889 (2013).
Kong, K. et al. An enantioselective total synthesis and stereochemical revision of (+)-citrinadin B. J. Am. Chem. Soc. 135, 10890–10893 (2013).
Bian, Z., Marvin, C. C., Pettersson, M. & Martin, S. F. Enantioselective total syntheses of citrinadins A and B. Stereochemical revision of their assigned structures. J. Am. Chem. Soc. 136, 14184–14192 (2014).
Matsumaru, T. et al. Synthetic studies toward the citrinadins: enantioselective preparation of an advanced spirooxindole intermediate. Tetrahedron 70, 4089–4093 (2014).
Smith, G. S. Progress Toward the Total Synthesis of the Citrinadins (Ph.D. Thesis Colorado State University, Fort Collins, CO, 2012).
Nakamura, R., Tanino, K. & Miyashita, M. Stereoselective synthesis of premisakinolide A, the monomeric counterpart of the marine 40-membered dimeric macrolide misakinolide A. Org. Lett. 7, 2929–2932 (2005).
Chackalamannil, S. & Wang, Y. An enantioselective route to trans-2,6-disubstituted piperidines. Tetrahedron 53, 11203 (1997).
Corey, E. J. & Chaykovsky, M. Dimethyloxosulfonium methylide ((CH3 2SOCH2 and dimethylsulfonium methylide ((CH3 2SCH2. Formation and application to organic synthesis. J. Am. Chem. Soc. 87, 1353–1364 (1965).
Aggarwal, V. K. & Winn, C. L. Catalytic, asymmetric sulfur ylide-mediated epoxidation of carbonyl compounds: scope, selectivity, and applications in synthesis. Acc. Chem. Res. 37, 611–620 (2004).
Caputo, R., Mangoni, L., Neri, O. & Palumbo, G. Direct conversion of oxiranes to alkenes by chlorotrimethylsilane and sodium iodide. Tetrahedron Lett. 22, 3551–3552 (1981).
Acknowledgements
We thank Amgen, Bristol-Myers Squibb and the NSF (CHE-1058292) for their financial support. We are grateful for generous funding from Baylor University, the Welch Foundation (Chair, AA-006) and the Cancer Research & Prevention Institute of Texas (CPRIT, R1309). MEM is grateful for the support of the NSF GRFP (2013156410). TM is grateful to the Uehara Foundation; Professor Toshiaki Sunazuka, Professor Satoshi Ōmura and the Kitasato Institute for postdoctoral support. JAE thanks the NIH (GM095076) for a postdoctoral fellowship. Dr Chris Rithner, Don Heyse, and Don Dick are acknowledged for their assistance in obtaining spectroscopic data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This manuscript is dedicated to Professor Amos B. Smith, III, an inspirational mentor and good friend.
Rights and permissions
About this article

Cite this article
McCallum, M., Smith, G., Matsumaru, T. et al. Synthetic studies toward citrinadin A: construction of the pentacyclic core. J Antibiot 69, 331–336 (2016). https://doi.org/10.1038/ja.2016.25
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.25
