
Abstract
Unified synthesis of FD-891 analogs and their structure–activity relationship are described. By using stereoselective allylation/crotylation and Evans aldol chemistry, six side-chain fragments having different length and terminus were synthesized. These fragments were coupled with a macrolactone fragment, improved synthesis of which was also developed here, to generate FD-891 and five truncated analogs. These synthetic compounds as well as three analogs obtained from fermentation of gene-disrupted Streptomyces graminofaciens mutants were tested for in vitro cytotoxic activity against HeLa cells. As a result, coexistence of the C8–C9 epoxide and side-chain terminus was found to be critical for the cytotoxic activity.
Similar content being viewed by others


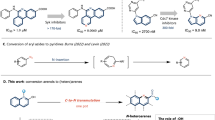
Introduction
FD-891 (1) and FD-892 (2) are biogenetically related macrolides isolated from the fermentation broth of Streptomyces graminofaciens A-8890 in 1994.1 These planar structures were first proposed as 18-membered macrolides,2 but were later reassigned as 16-membered compounds as shown in Figure 1.3 These macrolides have been reported to exhibit various biological activities.1, 4, 5 Among them, FD-891 (1) has been shown to have significant in vitro cytotoxicity against several tumor cell lines in the 10 nanomolar concentration range, whereas the level of cytotoxicity of FD-892 (2) was 100-fold lower.1 There are three structural differences between these molecules; that is, FD-891 (1) possesses a C8–C9 epoxide, C10-hydroxyl group and C25 methyl ether, all of which are tailored by post-polyketide synthase enzymes,6, 7 whereas none of these functional groups are found in FD-892 (2). Although three total syntheses8, 9, 10 and the preliminary structure–activity relationship data11 of FD-891 (1) have been reported, the detailed mode of action of FD-891 (1) as well as the origin of the difference in cytotoxic activity between FD-891 (1) and FD-892 (2) remains unknown.

FD-891 and FD-892.
We have been interested in the chemistry, biology and biogenesis of FD-891 (1). As part of this research, we herein report the unified synthesis of FD-891 analogs, and the structure–activity relationship of these analogs against human cervical carcinoma HeLa cells.
Results and discussion
Recently, we reported a concise and unified strategy for synthesis of the C1–C18 macrolactone fragments of FD-891 and 892 (3, 6) and their analogs (4, 5) (Figure 2).12 FD-891 macrolactone (3) possesses C1–C5 unsaturated ester and C8–C9 epoxide, both of which are thought to be representative protein-reactive functional groups.13 We speculated that the macrolactone portion is the minimal pharmacophore for cytotoxicity, and thus tested the synthesized macrolactones 3–6 for their in vitro cytotoxicity against HeLa cells. However, contrary to our expectation, none of the macrolactones—including compound 3, which has the correct C7–C10 oxygen functionality—exerted cytotoxic activity (IC50 (half-maximal inhibitory concentration) values: >60 μm) comparable to that of FD-891 (1, 8.1 nm).

Synthetic macrolactones 3–6.
These results indicated that the side-chain portion was necessary for FD-891 (1) to exert cytotoxic activity. To determine what functionality and length of the side-chain portion is needed for the biological activity, five truncated derivatives were designed as shown in Scheme 1; that is, the side-chain terminus was deleted in a stepwise manner to generate truncated derivatives 7–11. We anticipated that FD-891 (1) and its truncated derivatives 7–11 would be synthesized via Julia–Kocienski coupling of macrolactone aldehyde 12 and 1-phenyl-1H-tetrazol-5-yl (PT) sulfones 13–18, respectively.
The shortest C19–C21 side-chain fragment 18 was prepared according to the reported procedure14 with some modifications. The other fragments 13–17 were synthesized as shown in Scheme 2. To prepare these fragments, we set known aldehyde 1915, 16 as a common starting material. Brown crotylboration17 of 19 using (+)-Ipc2BOMe and alcohol protection using tert-butyldimethylsilyl chloride (TBSCl) or p-methoxybenzyl chloride (PMBCl) afforded TBS ether 21 and PMB ether 22, respectively. For synthesis of the C19–C23 side-chain fragment 17, ozonolysis of TBS ether 21 followed by NaBH4 reduction and protection of the resultant primary alcohol16 gave bis silylether 24 in 75% yield over three steps. Hydrogenolysis of benzyl ether, Mitsunobu coupling with 1-phenyl-1H-tetrazole-5-thiol and oxidation of the resultant thioether afforded PT sulfone 17 in 40% yield over three steps. To make the C19–C25 side-chain fragment 16 having no methyl group at C24, Lemieux–Johnson oxidation of PMB ether 22 followed by chelation-controlled stereoseletive allylation using allyltributyltin and MgBr218 gave homoallyl alcohol 25 in 55% yield over two steps. Anti-stereochemistry of the 1,3-diol moiety in 25 was confirmed by 13C-NMR after converted to the corresponding acetonide.19 The subsequent 8-step procedure including oxidative cleavage of terminal olefin and introduction of PT sulfone afforded the side-chain fragment 16 in 29% overall yield.
We expected that C19–C25 fragment 14 and its 25-O-demethyl analog 15 could be synthesized using crotylboration of aldehyde 23. However, it turned out to be difficult to obtain the desired stereoisomer selectively by crotylboration.20 We therefore used Evans aldol chemistry and obtained the coupling product 28 in 91% yield and good diastereoselectivity. After silylation of the secondary alcohol and subsequent LiBH4 reduction, protection (silylation or methylation) of the resultant primary alcohol gave silylether 30 and methylether 31 in 59% and 56% combined yields, respectively. Both compounds were successfully converted to the respective PT sulfones 15 and 14 in good overall yields. Fully elaborated C19–C26 fragment 13 was obtained in eight steps from intermediate 29, including diastereoselective reduction11 of methyl ketone 32.
Before coupling of these side-chain fragments with macrolactone 12, the synthetic scheme of 12 was optimized (Scheme 3). Starting from enal 34,12 organocatalytic allylation using chiral phosphoric acid 3521 developed by Jain and Antilla afforded homoallyl alcohol 36 in 94% yield and 93% ee. Previously, we used Brown allylboration, which requires stoichiometric and air/moisture-sensitive chiral auxiliary, to construct the C10 stereogenic center in our synthesis. Compared with that method, the present Antilla allylboration provides improvements in both catalytic efficiency and practicality. By using our reported 4-step sequence12 including vinylogous Mukaiyama aldol reaction using N,O-ketene silyl acetal 37,22 homoallyl alcohol 36 was converted to amide 38 as a single diastereomer in 86% overall yield. We previously protected the C7 alcohol of 38 as its TBS ether, but selective removal of this TBS group, which was crucial for stereoselective C8–C9 α-epoxidation,12 in the presence of C10 and C18 TBDPS ethers was difficult, and thus global deprotection followed by C10- and C18-selective protection had to be carried out. In the present case, we selected a more labile TES group for the protection of C7 alcohol, and the amide group of the resulting compound was reduced by diisobutylaluminum hydride (DIBAL-H) to give enal 39 in 87% yield over two steps. Wittig homologation of 39 followed by TMSOK (potassium trimethylsilanolate) treatment of the resulting methyl ester afforded α,β,γ,δ-unsaturated carboxylic acid 41 in 89% overall yield. Coupling of 41 with alcohol 42 using 2-methyl-6-nitrobenzoic anhydride (MNBA) proceeded smoothly under reflux temperature of CH2Cl2 to give the desired coupling product in 88% yield. The requested C7-selective deprotection was achieved by using catalytic PPTS (pyridinium p-toluenesulfonate) to afford allyl alcohol 43 in 92% yield. Although Sharpless epoxidation of 43 afforded a 4:1 inseparable mixture of the desired epoxy alcohol 44 and its diastereomer 45 in 70% combined yield, deprotection of the silyl groups followed by ring-closing metathesis and silica gel separation gave the desired ring-closing metathesis product 46 in 55% yield over two steps. The present route enabled the synthesis of 46 in 19% yield from enal 34, which is more than double the yield compared with the previous route12 (9% overall yield). Triol 46 was converted to aldehyde 12 via a 3-step sequence that included oxidation using AZADO (2-azaadamantane N-oxyl) and CuCl under an O2 atmosphere.23 Finally, Julia–Kocienski coupling of aldehyde 12 and PT sulfones 13–18 followed by global deprotection afforded FD-891 (1) and its side-chain-truncated derivatives 7–11, as expected.
Having the natural product and its truncated derivatives in hand, we next evaluated the in vitro cytotoxic activity of these compounds against HeLa cells. In addition to the synthetic samples, compounds obtained by fermentation of gene-disrupted S. graminofaciens A-8890 mutants6, 7 were also evaluated. These results are listed in Table 1.
25-O-Demethyl-FD-891 (47)7 exhibited a fivefold lower level of activity compared with FD-891 (1), whereas the cytotoxic activity of 10-deoxy-FD-891 (48)6 was almost the same as that of FD-891 (1). These data showed that the presence of C10-hydroxyl group had almost no or even a negative impact on the cytotoxicity, but the C25-O-methyl group seemed to enhance the cytotoxic activity. In sharp contrast to these functional groups, C8–C9 epoxide was found to be critical, as 25-O-methyl-FD-892 (49)6 was more than 800 times less active than FD-891 (1). This result strongly indicated that the coexistence of the side-chain portion and C8–C9 epoxide was crucial because macrolactone 3 was found to be inactive (vide supra). Interestingly, removal of O-methyl from the side-chain terminus had a smaller effect than removal of C-methyl (i.e., 47 vs 7). We speculated that the length of the carbon side chain was very important. Further removal of C25-O-methyl (8) and C24-methyl (9) did not affect the compound's activity, whereas the more truncated derivatives 10 and 11 lost the cytotoxic activity in a stepwise manner.
In summary, we have synthesized FD-891 (1) and five analogs having a truncated side chain, and carried out a structure–activity relationship study of FD-891 (1) by using nine synthetic compounds and three analogs obtained by the fermentation of gene-disrupted S. graminofaciens mutants. These compounds were evaluated for their in vitro cytotoxic activity against HeLa cells, and the presence of both the side chain and C8–C9 epoxide was found to be critical for the cytotoxicity. Moreover, the length of the side chain and environment of its terminus were also important. It is worth noting that C25 O-methylation and P450-catalyzed C8–C9 epoxidation, two of the three steps of post-polyketide synthase modification in FD-891 biosynthesis,6 greatly enhanced the compound's activity, whereas C10 hydroxylation did not improve the activity of the compound. Although the exact role(s) of these functional groups remain to be clarified, the results and derivatives obtained in this study would be useful for further mode-of-action studies. The results obtained from these studies will be reported in due course.
Experimental procedures
All reactions were carried out under an argon atmosphere with dehydrated solvents under anhydrous conditions, unless otherwise noted. Dehydrated THF and CH2Cl2 were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). Other solvents were dehydrated and distilled according to standard protocols. Reagents were obtained from commercial suppliers and used without further purification, unless otherwise noted. Reactions were monitored by TLC carried out on E Merck Silica gel 60 F254 precoated plates (Merck, Frankfurt, Germany). Column chromatography was performed on Silica gel 60N (spherical, neutral, 63–210 μm; Kanto Chemical Co., Ltd.) and flash column chromatography was performed on Silica gel 60N (spherical, neutral, 40–50 μm; Kanto Chemical Co., Ltd.). Optical rotations were measured on a JASCO DIP-370 Digital Polarimeter or JASCO P-2200 Digital Polarimeter (JASCO Corporation, Tokyo, Japan) at room temperature, using the sodium D line. IR spectra were recorded on a JASCO FT/IR-410 Fourier Transform Infrared Spectrophotometer (JASCO Corporation). 1H-NMR (400 and 600 MHz) and 13C-NMR spectra (100 and 150 MHz) were recorded on JEOL JNM-AL-400 and JEOL JNM-ECA-600 spectrometers, respectively (JEOL Resonance Inc., Tokyo, Japan). For 1H-NMR spectra, chemical shifts (δ) are given from TMS (0.00 p.p.m.) in CDCl3 as internal standards. For 13C-NMR spectra, chemical shifts (δ) are given from CDCl3 (77.0 p.p.m.) as internal standards. The following abbreviations were used to explain the multiplicities: s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, sept=septet and br=broad. Mass spectra were recorded on a JEOL JMS-DX303, JEOL JNM-AL500 and JEOL JMS-700 (JEOL Ltd., Tokyo, Japan).

Design and synthetic strategy of side-chain-truncated derivatives 7–11. PT=1-phenyl-1H-tetrazol-5-yl; Si=tert-butyldimethylsilyl.

Synthesis of side-chain fragments 13–17. Reagents and conditions: (a) cis-2-butene, tert-BuOK, n-BuLi, (+)-Ipc2BOMe, BF3·OEt2, THF (tetrahydrofuran), −78 °C, then NaOH, aq. H2O2, MeOH, room temperature (rt); (b) TBSCl, imidazole, DMF (dimethylformamide), 0 °C to rt; (c) PMBCl, NaH, DMF, 0 °C to rt; (d) O3, MeOH-CH2Cl2, −78 °C, then PPh3; (e) NaBH4, MeOH, 0 °C; (f) TBSCl, imidazole, CH2Cl2, 0 °C; (g) OsO4, NaIO4, 2,6-lutidine, dioxane-H2O, rt; (h) allyltributyltin, MgBr2·OEt2, CH2Cl2, rt; (i) TFA, CH2Cl2, 0 °C; (j) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C; (k) oxazolidinone 27, i-Pr2NEt, CH2Cl2, −78 °C to −50 °C; (l) LiBH4, Et2O-EtOH, 0 °C to rt; (m) MeOTf, proton sponge, CH2Cl2, 0 °C to rt; (n) LiOH, aq. H2O2, THF, 0°C to rt; (o) NH(OMe)Me·HCl, CDI, CH2Cl2, rt; (p) MeMgBr, THF, −20 °C to rt; (q) Me2AlCl, Bu3SnH, CH2Cl2, −90 °C; (r) MeOTf, proton sponge, CHCl3, reflux; (s) H2, Pd/C, EtOH, rt; (t) H2, Pd(OH)2/C, EtOH, rt; (u) 1-phenyl-1H-tetrazole-5-thiol, Ph3P, DIAD, THF, 0 °C to rt; (v) (NH4)6Mo7O24·4H2O, aq. H2O2, EtOH, 0 °C to rt. Si=tert-butyldimethylsilyl.

Synthesis of FD-891 (1) and its side-chain-truncated derivatives 7–11. Reagents and conditions: (a) cat. (R)-TRIP-PA 35, allylpinacolborane, toluene, −40 °C; (b) TESOTf, 2,6-lutidine, CH2Cl2, 0 °C; (c) DIBAL-H, CH2Cl2, −78 °C; (d) Wittig reagent 40, toluene, reflux; (e) TMSOK, THF, rt; (f) MNBA, Et3N, DMAP, CH2Cl2, reflux; (g) PPTS, MeOH, rt; (h) Ti(iOPr)4, (+)-DIPT, TBHP, MS4A, CH2Cl2, −25 °C; (i) TBAF, THF, 40 °C; (j) Grubbs second-generation catalyst, tetrafluoro-1,4-benzoquinone, CH2Cl2, reflux; (k) TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C to rt; (l) PPTS, MeOH, rt; (m) AZADO, CuCl, bpy, DMAP, CH3CN, O2, rt; (n) sulfone, KHMDS, DME, −60 °C; (o) H2SiF6, H2O-CH3CN, rt.
General procedure for the synthesis of PT sulfones: synthesis of PT sulfone 13
To a solution of precursor sulfide (69.9 mg, 0.115 mmol) in EtOH (5.80 ml) was added (NH4)6Mo7O24·4H2O (42.5 mg, 34.4 μmol) followed by H2O2 aq. (0.13 ml, 8.84 m, 1.15 mmol) at 0 °C. After stirring for 15 h at room temperature, the reaction mixture was quenched with saturated Na2S2O3 aq. (10.0 ml) and extracted with EtOAc. The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/hexane=1/10) to give sulfone (62.7 mg, 97.8 μmol, 85%) as a colorless oil.
13: [α]22D +6.4 (c 0.72, CHCl3); IR (neat): 2929 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.73–7.70 (m, 2H), 7.63–7.57 (m, 3H), 3.89–3.76 (m, 4H), 3.26 (s, 3H), 3.19–3.16 (m, 1H), 2.40–2.31 (m, 1H), 2.21–2.12 (m, 1H), 1.78–1.70 (m, 1H), 1.68–1.61 (m, 1H), 1.12 (d, J=6.0 Hz, 3H), 0.96 (d, J=6.8 Hz, 3H), 0.93 (d, J=6.8 Hz, 3H), 0.91 (s, 9H), 0.90 (s, 9H), 0.110 (s, 3H), 0.107 (s, 3H), 0.095 (s, 3H), 0.08 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 153.5, 133.1, 131.4, 129.7, 125.0, 79.6, 72.0, 71.1, 56.2, 51.9, 43.9, 41.2, 27.2, 26.0, 25.9, 18.4, 18.1, 15.7, 12.5, 11.4, −3.3, −4.1, −4.27, −4.33; HR-MS (ESI) calcd. for C30H57N4O5SSi2 [M++H] 641.3583, found 641.3568.
PT sulfone 14
[α]19D +1.2 (c 0.80, CHCl3); IR (neat): 2955 cm−1; 1H-NMR (600 MHz, CDCl3) δ 7.72–7.70 (m, 2H), 7.64–7.58 (m, 3H), 3.94–3.93 (m, 1H), 3.85–3.76 (m, 3H), 3.28 (s, 3H), 3.19 (dd, J=8.7, 8.7 Hz, 1H), 3.10 (dd, J=9.0, 5.4 Hz, 1H), 2.36–2.30 (m, 1H), 2.16–2.10 (m, 1H), 1.79–1.73 (m, 1H), 1.73–1.67 (m, 1H), 0.98 (d, J=7.2 Hz, 3H), 0.912 (s, 9H), 0.906 (s, 9H), 0.86 (d, J=7.2 Hz, 3H), 0.11–0.08 (m, 12H); +++++13C-NMR (150 MHz, CDCl3) δ 153.4, 133.1, 131.4, 129.7, 125.0, 76.4, 71.6, 70.9, 58.6, 51.7, 43.9, 35.5, 29.7, 27.1, 26.0, 25.9, 18.3, 18.1, 12.4, 11.7, −3.8, −4.1, −4.4, −4.6; LR-MS (EI) m/z 569 (M+-tBu), 117 (100%); HR-MS (EI) calcd. for C25H45N4O5SSi2 (M+-tBu) 569.2644, found 569.2669.
PT sulfone 15
[α]18D −0.30 (c 0.31, CHCl3); IR (neat): 2928 cm−1; 1H-NMR (600 MHz, CDCl3) δ 7.70 (dd, J=8.4, 1.8 Hz, 2H), 7.62–7.59 (m, 3H), 3.88 (m, 1H), 3.87–3.84 (m, 1H), 3.82–3.80 (m, 2H), 3.44 (dd, J=9.6, 8.2 Hz, 1H), 3.37 (dd, J=9.6, 6.6 Hz, 1H), 2.31–2.25 (m, 1H), 2.18–2.12 (m, 1H), 1.73–1.68 (m, 2H), 0.94 (d, J=7.2 Hz, 3H), 0.903 (s, 9H), 0.899 (s, 9H), 0.87 (s, 9H), 0.84 (d, J=6.6 Hz, 3H), 0.11–0.09 (m, 12H), 0.02–0.01 (m, 6H); 13C-NMR (150 MHz, CDCl3) δ 153.5, 131.4, 129.7, 125.0, 71.6, 70.9, 66.6, 51.8, 43.5, 38.3, 29.7, 27.4, 26.02, 25.96, 25.9, 18.4, 18.1, 12.3, 11.3, −3.6, −4.1, −4.3, −4.4, −5.3, −5.4; LR-MS (EI) m/z 669 (M+-tBu), 117 (100%); HR-MS (EI) calcd. for C30H57N4O5SSi3 (M+-tBu) 669.3352, found 669.3357.
PT sulfone 16
[α]24D −7.5 (c 0.72, CHCl3); IR (neat): 2954 cm-1; 1H-NMR (400 MHz, CDCl3) δ 7.72–7.70 (m, 2H), 7.65–7.57 (m, 3H), 3.93–3.89 (m, 1H), 3.83 (dd, J=9.6, 7.2 Hz, 2H), 3.76–3.71 (m, 2H), 3.64 (dt, J=9.6, 7.2 Hz, 1H), 2.21–2.15 (m, 2H), 1.74–1.69 (m, 1H), 1.65–1.57 (m, 1H), 1.51–1.43 (m, 1H), 0.95 (d, J=6.8 Hz, 3H), 0.91 (s, 9H), 0.89 (s, 9H), 0.87 (s, 9H), 0.11 (s, 3H), 0.10 (m, 6H), 0.08 (s, 3H), 0.04 (s, 6H); 13C-NMR (100 MHz, CDCl3) δ 153.5, 133.1, 131.4, 129.7, 125.0, 71.6, 69.2, 60.3, 51.7, 43.4, 35.3, 26.9, 26.0, 25.9, 25.8, 18.3, 18.1, 18.0, 11.0, −4.18, −4.22, −4.5, −4.6, −5.3; HR-MS (ESI) calcd. for C33H65N4O5SSi3 (M++H) 713.3978, found 713.3974.
PT sulfone 17
[α]27D +3.40 (c 1.22, CHCl3); IR (neat): 2929 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.71–7.69 (m, 2H), 7.63–7.57 (m, 3H), 3.91 (q, J=5.4 Hz, 1H), 3.83 (ddd, J=14.6, 11.2, 5.9 Hz, 1H), 3.74 (ddd, J=14.1, 10.7, 5.4 Hz, 1H), 3.56–3.49 (m, 2H), 2.20–2.07 (m, 2H), 1.72–1.66 (m, 1H), 0.91–0.87 (m, 21H), 0.08 (s, 3H), 0.07 (s, 3H), 0.03 (s, 6H); 13C-NMR (100 MHz, CDCl3): δ 153.5, 133.1, 131.4, 129.7, 125.0, 71.1, 64.3, 53.1, 40.6, 26.6, 25.9, 25.8, 18.2, 18.1, 12.2, −4.4, −4.6, −5.4, −5.5; LR-MS (FAB) m/z 555.3 (M++H), 73.1 (100%); HR-MS (FAB) calcd. for C25H47N4O4SSi2 (M++H) 555.2851, found 555.2859.
Synthesis of aldehyde 12
To a solution of precursor alcohol (23.0 mg, 37.0 μmol), AZADO (0.56 mg, 3.7 μmol), bpy (0.58 mg, 3.7 μmol) and DMAP (0.90 mg, 7.40 μmol) in MeCN (1.90 ml) was added CuCl (0.73 mg, 7.4 μmol) at room temperature. After stirring for 15 min under an O2 atmosphere, the reaction mixture was quenched with saturated NaHCO3 aq. (5.0 ml) and extracted with EtOAc. The combined organic layers were dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/hexane=1/10) to give aldehyde (20.9 mg, 33.6 μmol, 91%) as a colorless oil. The aldehyde was immediately used for the next step.
General procedure for the synthesis of FD-891 and its truncated analogs: synthesis of FD-891 (1)
To a solution of sulfone 13 (30.1 mg, 46.9 μmol) and crude aldehyde (8.85 mg, 14.3 μmol) in DME (1.4 ml) was added KHMDS (94 μl, 0.5 m solution in toluene, 46.9 μmol) at −60 °C. After stirring for 20 min, the reaction mixture was quenched with saturated NH4Cl aq. (3.0 ml) and extracted with EtOAc. The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/hexane=1/20) to give coupling adduct (8.11 mg, 7.83 μmol, 55%) as a colorless oil.
To a solution of coupling adduct (5.19 mg, 5.01 μmol) in MeCN (0.6 ml) was added fluorosilic acid 40–45 wt% solution in water (0.100 ml) at room temperature. After stirring for 4 h, the reaction mixture was quenched with saturated NaHCO3 aq. (5.0 ml) and extracted with EtOAc. The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc) to give FD-891 (1) (1.25 mg, 2.16 μmol, 43%) as a colorless oil.
1: [α]22D +4.9 (c 0.080, MeOH); IR (neat): 3446 (br), 1697 cm−1; 1H-NMR (600 MHz, CDCl3): δ 7.30 (s, 1H), 5.77 (ddd, J=15.0, 10.2, 4.8 Hz, 1H), 5.63–5.49 (m, 4H), 4.84 (dt, J=9.6, 3.6 Hz, 1H), 4.15 (brs, 1H), 4.00 (s, 1H), 3.86 (brs, 1H), 3.82 (dd, J=9.6, 1.2 Hz, 1H), 3.62–3.58 (m, 2H), 3.34 (s, 3H), 3.25 (dd, J=4.8, 3.0 Hz, 1H), 3.16 (dd, J=1.8, 1.8 Hz, 1H), 3.12 (ddd, J=11.4, 7.8, 3.6 Hz, 1H), 2.87 (brs, 1H), 2.56–2.45 (m, 3H), 2.33–2.17 (m, 4H), 2.09 (s, 3H), 2.03 (s, 3H), 2.01–1.94 (m, 2H), 1.91–1.81(m, 3H), 1.19 (d, J=6.0 Hz, 3H), 1.14 (d, J=7.8 Hz, 3H), 0.93 (d, J=7.2 Hz, 3H), 0.91 (d, J=6.6 Hz, 3H), 0.79 (d, J=7.8 Hz, 3H); 13C-NMR (150 MHz, CDCl3): δ 168.8, 144.0, 141.7, 135.8, 130.2, 129.4, 129.3, 128.1, 124.7, 82.7, 78.4, 76.5, 73.2, 71.0, 70.9, 56.0, 55.9, 55.0, 39.7, 39.6, 38.2, 36.8, 36.0, 35.9, 34.5, 34.4, 16.5, 16.4, 16.3, 15.6, 13.7, 11.6, 4.9; HR-MS (ESI) calcd. for C33H55O8 [M++H] 579.3891, found 579.3878.
Truncated analog 7
[α]28D −26 (c 0.080, CHCl3); IR (neat): 3421 (br), 1696 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.30 (s, 1H), 5.76 (ddd, J=15.2, 9.6, 5.2 Hz, 1H), 5.62–5.48 (m, 4H), 4.84 (dt, J=9.2, 3.6 Hz, 1H), 4.15 (brs, 1H), 3.88 (d, J=8.8 Hz, 1H), 3.82 (dd, J=9.6, 2.0 Hz, 1H), 3.60 (brs, 1H), 3.47 (d, J=4.8 Hz, 2H), 3.37–3.35 (m, 4H), 3.25 (dd, J=4.8, 2.4 Hz, 1H), 3.15 (dd, J=2.0, 2.0 Hz, 1H), 3.14–3.10 (m, 1H), 2.72 (brs, 1H), 2.56–2.28 (m, 3H), 2.27–2.17 (m, 4H), 2.09 (s, 3H), 2.02 (s, 3H), 1.99–1.82 (m, 6H), 1.14 (d, J=7.2 Hz, 3H), 0.97 (d, J=7.2 Hz, 3H), 0.91 (d, J=6.4 Hz, 3H), 0.83 (d, J=7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3): δ 168.8, 144.0, 141.7, 135.8, 130.0, 129.9, 129.2, 128.1, 124.6, 78.2, 76.4, 76.0, 73.4, 71.0, 70.9, 59.2, 55.9, 55.1, 39.3, 38.2, 36.6, 36.1, 35.9, 35.2, 34.54, 34.47, 16.5, 16.3, 15.6, 13.7, 11.7, 9.7; HR-MS (ESI) calcd. for C32H53O8 [M++H] 565.3735, found 565.3724.
Truncated analog 8
[α]28D −21 (c 0.23, CHCl3); IR (neat): 3407 (br), 1680 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.30 (s, 1H), 5.76 (ddd, J=15.2, 9.6, 5.2 Hz, 1H), 5.63–5.48 (m, 4H), 4.85 (dt, J=9.2, 3.6 Hz, 1H), 4.15 (brs, 1H), 3.91–3.88 (m, 2H), 3.79 (dd, J=10.8, 4.0 Hz, 1H), 3.71 (dd, J=10.8, 5.6 Hz, 1H), 3.65–3.60 (m, 2H), 3.24 (dd, J=4.8, 2.8 Hz, 1H), 3.15 (dd, J=2.0, 2.0 Hz, 1H), 3.13–3.10 (m, 1H), 2.56–2.43 (m, 4H), 2.33–2.17 (m, 5H), 2.08 (s, 3H), 2.02 (s, 3H), 2.00–1.90 (m, 5H), 1.80–1.74 (m, 1H), 1.13 (d, J=7.2 Hz, 3H), 0.99 (d, J=7.2 Hz, 3H), 0.91 (d, J=7.2 Hz, 3H), 0.83 (d, J=7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3): δ 168.9, 144.2, 141.7, 135.8, 130.9, 129.4, 129.1, 128.3, 124.5, 76.2, 76.1, 73.7, 71.0, 70.9, 67.9, 55.9, 55.1, 39.1, 38.2, 36.4, 36.1, 35.9, 34.6, 34.5, 16.5, 16.3, 15.6, 13.6, 11.8, 9.1; HR-MS (ESI) calcd. for C31H51O8 [M++H] 551.3578, found 551.3569.
Truncated analog 9
[α]27D −20 (c 0.12, CHCl3); IR (neat): 3389 (br), 1693 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.30 (s, 1H), 5.76 (ddd, J=15.2, 9.6, 4.8 Hz, 1H), 5.63–5.45 (m, 4H), 4.85 (dt, J=9.2, 3.6 Hz, 1H), 4.15 (brs, 1H), 3.99–3.84 (m, 5H), 3.60 (brs, 1H), 3.24 (dd, J=4.4, 2.8 Hz, 1H), 3.15 (dd, J=2.4, 2.0 Hz, 1H), 3.14–3.10 (m, 1H), 2.70 (brs, 1H), 2.56–2.43 (m, 3H), 2.32–2.18 (m, 4H), 2.08 (s, 3H), 2.02 (s, 3H), 1.99–1.82 (m, 5H), 1.74–1.69 (m, 2H), 1.14 (d, J=6.8 Hz, 3H), 0.97 (d, J=7.2 Hz, 3H), 0.91 (d, J=6.4 Hz, 3H); 13C-NMR (100 MHz, CDCl3): δ 169.0, 144.2, 141.8, 135.8, 130.9, 129.1, 129.0, 128.3, 124.5, 76.3, 76.2, 72.4, 71.0, 70.9, 62.2, 55.9, 55.1, 41.4, 38.2, 37.1, 36.6, 36.1, 35.9, 34.54, 34.50, 16.5, 16.3, 15.6, 13.7, 11.4; HR-MS (ESI) calcd. for C30H49O8 [M++H] 537.3422, found 537.3413.
Truncated analog 10
[α]26D −8.3 (c 0.11, CHCl3); IR (neat): 3406 (br), 1693 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.30 (s, 1H), 5.76 (ddd, J=15.2, 10.0, 5.2 Hz, 1H), 5.63–5.46 (m, 4H), 4.85 (dt, J=9.2, 3.6 Hz, 1H), 4.15 (brs, 1H), 3.85 (dt, J=6.8, 2.8 Hz, 1H), 3.71 (d, J=4.8 Hz, 2H), 3.60 (brs, 1H), 3.24 (dd, J=4.4, 2.4 Hz, 1H), 3.15 (dd, J=2.4, 2.0 Hz, 1H), 3.14–3.10 (m, 1H), 2.56–2.42 (m, 3H), 2.25–2.18 (m, 6H), 2.09 (s, 3H), 2.04–1.84 (m, 8H), 1.14 (d, J=6.8 Hz, 3H), 0.95 (d, J=6.8 Hz, 3H), 0.91 (d, J=6.4 Hz, 3H); 13C-NMR (100 MHz, CDCl3): δ 168.9, 144.1, 141.7, 135.8, 131.0, 129.1, 128.9, 128.3, 124.6, 76.2, 73.7, 71.0, 70.9, 67.0, 55.9, 55.1, 38.8, 38.2, 37.6, 36.1, 35.9, 34.5, 16.5, 16.3, 15.6, 13.7, 10.3; HR-MS (ESI) calcd. for C28H45O7 [M++H] 493.3160, found 493.3153.
Truncated analog 11
[α]25D −13 (c 0.12, CHCl3); IR (neat): 3414 (br), 1696 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.30 (s, 1H), 5.76 (ddd, J=15.2, 10.0, 5.2 Hz, 1H), 5.63–5.43 (m, 4H), 4.85 (dt, J=9.2, 3.6 Hz, 1H), 4.15 (brs, 1H), 3.66 (t, J=6.0 Hz, 2H), 3.60 (brs, 1H), 3.24 (dd, J=4.8, 2.4 Hz, 1H), 3.15 (dd, J=2.4, 2.0 Hz, 1H), 3.14–3.08 (m, 1H), 2.60–2.42 (m, 3H), 2.33–2.18 (m, 4H), 2.09 (s, 3H), 2.02–1.86 (m, 8H), 1.14 (d, J=7.2 Hz, 3H), 0.91 (d, J=6.4 Hz, 3H); 13C-NMR (100 MHz, CDCl3): δ 168.9, 144.0, 141.7, 135.8, 130.7, 129.1, 128.7, 128.2, 124.6, 76.2, 71.0, 70.9, 62.1, 55.9, 55.1, 38.2, 36.10, 36.06, 35.9, 34.53, 34.49, 16.5, 16.2, 15.6, 13.7; HR-MS (ESI) calcd. for C25H39O6 [M++H] 435.2741, found 435.2735.
In vitro cytotoxic assay
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (Nichirei Biosciences, Tokyo, Japan) in a humidified atmosphere containing 5% CO2. For viability assay, HeLa cells were seeded at 3 × 103 cells per 100 μl per well in a 96-well microplate, and treated with various concentrations of FD-891 derivatives for 48 h. IC50 values were determined using the cell counting reagent WST-8 (Dojindo, Kumamoto, Japan).
References
Seki-Asano, M. et al. Isolation and characterization of new 18-membered macrolides FD-891 and FD-892. J. Antibiot. 47, 1226–1233 (1994).
Seki-Asano, M., Tsuchida, Y., Hanada, K. & Mizoue, K. Structures of new 18-membered macrolides FD-891 and FD-892. J. Antibiot. 47, 1234–1241 (1994).
Eguchi, T., Yamamoto, K., Mizoue, K. & Kakinuma, K. Structure revision of FD-891, a 16-membered macrolide antibiotic. J. Antibiot. 57, 156–157 (2004).
Kataoka, T. et al. FD-891, a structural analogue of concanamycin A that does not affect vacuolar acidification or perforin activity, yet potently prevents cytotoxic T lymphocyte-mediated cytotoxicity through the blockage of conjugate formation. Immunology 100, 170–177 (2000).
Inaba, S. et al. The cytotoxic macrolide FD-891 induces caspase-8-dependent mitochondrial release of cytochrome c and subsequent apoptosis in human leukemia Jurkat cells. J. Antibiot. 62, 507–512 (2009).
Kudo, F., Motegi, A., Mizoue, K. & Eguchi, T. Cloning and characterization of the biosynthetic gene cluster of 16-membered macrolide antibiotic FD-891: involvement of a dual functional cytochrome P450 monooxygenase catalyzing epoxidation and hydroxylation. Chembiochem 11, 1574–1582 (2010).
Kudo, F. et al. Parallel post-polyketide synthase modification mechanism involved in FD-891 biosynthesis in Streptomyces graminofaciens A-8890. Chembiochem (e-pub ahead of print 2 December 2015; doi:10.1002/cbic.201500533).
Crimmins, M. T. & Caussanel, F. Enantioselective total synthesis of FD-891. J. Am. Chem. Soc. 128, 3128–3129 (2006).
Garcia-Fortanet, J., Murga, J., Carda, M. & Marco, J. A. Stereoselective synthesis of the cytotoxic macrolide FD-891. Org. Lett. 8, 2695–2698 (2006).
Yadav, J. S., Das, S. K. & Sabitha, G. Stereoselective total synthesis of FD-891. J. Org. Chem. 77, 11109–11118 (2012).
Garcia-Fortanet, J. et al. The total synthesis and biological properties of the cytotoxic macrolide FD-891 and its non-natural (Z-C12 isomer. Chem. Eur. J 13, 5060–5074 (2007).
Kanoh, N. et al. A concise and unified strategy for synthesis of the C1–C18 macrolactone fragments of FD-891, FD-892 and their analogues: formal total synthesis of FD-891. Org. Lett. 16, 5216–5219 (2014).
Drahl, C., Cravatt, F. & Sorensen, E. J. Protein-reactive natural products. Angew. Chem. Int. Ed. 44, 5788–5809 (2005).
Sedrani, R. et al. Sanglifehrin–cyclophilin interaction: degradation work, synthetic macrocyclic analogues, X-ray crystal structure, and binding data. J. Am. Chem. Soc. 125, 3849–3859 (2003).
Wakamiya, T., Konishi, K., Chaki, H., Teshima, T. & Shiba, T. Chemical studies on tuberactinomycin. XVIII. Syntheses of dl-dihydroviomycidine and dl-viomycidine. Heterocycles 15, 999–1005 (1981).
Chandrasekhar, B., Athe, S., Reddy, P. P. & Ghosh, S. Synthesis of fully functionalized aglycone of lycoperdinoside A and B. Org. Biomol. Chem. 13, 115–124 (2015).
Brown, H. C., Bhat, K. S. & Randad, R. S. Chiral synthesis via organoboranes. 21. Allylboration and crotylboration of alpha-chiral aldehydes with diisopinocampheylboron as the chiral auxiliary. J. Org. Chem. 54, 1570–1576 (1989).
Fuwa, H. & Sasaki, M. Total synthesis of (−)-exiguolide. Org. Lett. 12, 584–587 (2010).
Rychnovsky, S. D., Rogers, B. & Yang, G. Analysis of two 13C NMR correlations for determining the stereochemistry of 1,3-diol acetonides. J. Org. Chem. 58, 3511–3515 (1993).
Lee, J., Kobayashi, Y., Tezuka, K. & Kishi, Y. Toward creation of a universal NMR database for the stereochemical assignment of acyclic compounds: proof of concept. Org. Lett. 1, 2181–2184 (1999).
Jain, P. & Antilla, J. C. Chiral Brønsted acid-catalyzed allylboration of aldehydes. J. Am. Chem. Soc. 132, 11884–11886 (2010).
Shirokawa, S. I. et al. Remote asymmetric induction with vinyl ketene silyl N,O-acetal. J. Am. Chem. Soc. 126, 13604–13605 (2004).
Sasano, Y., Kogure, N., Nishiyama, T., Nagasawa, S. & Iwabuchi, Y. Highly efficient aerobic oxidation of alcohols by using less-hindered nitroxyl-radical/copper catalysis: optimum catalyst combinations and their substrate scope. Chem. Asian J. 10, 1004–1009 (2015).
Acknowledgements
We acknowledge Prof. Masahiro Terada and Mr. Takuto Yamanaka (Graduate School of Science, Tohoku University) for kindly providing information on the organocatalytic allylation using catalyst 35. This work was supported by a Grant-in-Aid for Scientific Research on the Innovative Area ‘Chemical Biology of Natural Products’ from The Ministry of Education, Culture, Sports, Science and Technology, Japan (No. 23102013 to NK), and a Research Fellowship Grant for Young Researchers from Japan Society for the Promotion of Science (15J03635 to AK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Dedicated to Professor Amos B. Smith, III in celebration of his 50 years of contributions to the chemical sciences.
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Itagaki, T., Kawamata, A., Takeuchi, M. et al. Synthesis and structure–activity relationship study of FD-891: importance of the side chain and C8–C9 epoxide for cytotoxic activity against cancer cells. J Antibiot 69, 287–293 (2016). https://doi.org/10.1038/ja.2015.148
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.148
This article is cited by
-
The Combinatorial Biosynthesis of “Unnatural” Products with Polyketides
Transactions of Tianjin University (2018)
