
Abstract
Resources structure ecological communities and potentially link biodiversity to energy flow. It is commonly believed that functional traits (generalists versus specialists) involved in the exploitation of resources depend on resource availability and environmental fluctuations. The longitudinal nature of stream ecosystems provides changing resources to stream biota with yet unknown effects on microbial functional traits and community structure. We investigated the impact of autochthonous (algal extract) and allochthonous (spruce extract) resources, as they change along alpine streams from above to below the treeline, on microbial diversity, community composition and functions of benthic biofilms. Combining bromodeoxyuridine labelling and 454 pyrosequencing, we showed that diversity was lower upstream than downstream of the treeline and that community composition changed along the altitudinal gradient. We also found that, especially for allochthonous resources, specialisation by biofilm bacteria increased along that same gradient. Our results suggest that in streams below the treeline biofilm diversity, specialisation and functioning are associated with increasing niche differentiation as potentially modulated by divers allochthonous and autochthonous constituents contributing to resources. These findings expand our current understanding on biofilm structure and function in alpine streams.
Similar content being viewed by others

Introduction
In headwater streams, the smallest and most abundant in stream networks, dissolved organic carbon (DOC) constitutes the primary resource for microbial heterotrophs. DOC encompasses a large pool of molecules, which depending on their source (i.e., autochthonous versus allochthonous), can vary in chemical composition and bioavailability (e.g., Sun et al., 1997; Jaffé et al., 2012). Allochthonous constituents typically dominate the DOC pool, with the exception of alpine and arid catchments devoid of major terrestrial vegetation (e.g., Milner et al., 2009). In alpine streams, autochthonous deliveries from algal production (and mosses) characterise DOC in streams above the treeline, whereas terrestrial deliveries increasingly add to the carbon pool further downstream (Battin et al., 2004; Milner et al., 2009; Zah and Uehlinger, 2001). Several studies have reported DOC source and composition as potential drivers of microbial community structure and functioning in streams (e.g., Kaplan and Bott, 1989; Sun et al., 1997; Peter et al., 2011) and in lakes (e.g., Crump et al., 2003; Judd et al., 2006). However, only few have studied the relationship between DOC characteristics and microbial functional traits in streams (McArthur et al., 1985; Koetsier et al., 1997). Although with a traditional focus on plants and animals, the concept of functional traits (i.e., specialists versus generalists) now becomes increasingly established in microbial ecology with notable examples from soil (Lennon et al., 2012), wastewater (Muller et al., 2014) and marine ecosystems (Mou et al., 2008). Understanding the relationship between microbial community structure and specialisation in response to resources may shed new light on the ecological mechanisms underlying the carbon cycling in stream ecosystems (Battin et al., 2008). However, trait distribution and the role of functional redundancy remain elusive in stream biofilms, especially when multiple functions are considered simultaneously (Gamfeldt et al., 2008; Hillebrand and Matthiessen, 2009).
The stochastic nature of hydrology and sedimentary dynamics in streams creates pronounced heterogeneity in resource availability and biodiversity patterns (Townsend, 1989). It is assumed that these environmental fluctuations relieve invertebrate species from competition, which would favour generalists rather than specialists (Townsend, 1989; Schoener, 1974; Rosenfeld, 2002). At the same time, it is established that resource partitioning allows invertebrates to assemble diverse communities along the stream continuum. The relationships between downstream shifts in resources (i.e., primarily autochthonous sources upstream versus more allochthonous sources downstream), hydro-geomorphology and biotic communities have been described by the River Continuum Concept (Vannote et al., 1980). Microorganisms were not encapsulated in the River Continuum Concept, and yet evidence increasingly suggests that microbial communities predictably change along the river continuum (Besemer et al., 2013; Read et al., 2014; Savio et al., 2014). However, the potential drivers of these patterns remain poorly understood, which is noticeable, given the relevance of microorganisms for stream ecosystem functions (Battin et al., 2008).
The aim of this study was to investigate microbial community structure, diversity and functional traits (generalists versus specialists) and redundancy in benthic biofilms of Alpine streams in response to resources that potentially change from upstream to downstream of the treeline. We hypothesised that microbial community structure and diversity change along such altitudinal gradients and that resource specialisation contributes to these patterns. Mixing of resources from various sources along the longitudinal continuum of streams may increase substrate diversity and reduce its relative dominance by averaging. Although microbial generalists may be able to better cope with pronounced environmental and resource fluctuations upstream of the treeline, we expect that microbial specialists increasingly establish downstream because of potentially increasing resource diversity. Hence, we postulated that the relative importance of microbial specialists versus generalists would increase downstream of the treeline where both autochthonous and allochthonous sources contribute to resource diversity and abundance. We further expected that microorganisms in reaches above the treeline would primarily use autochthonous DOC with only limited capacity to metabolise allochthonous DOC constituents; this would result in a mixture of microbial generalists and specialists for autochthonous DOC in these reaches. Streams below the treeline, on the other hand, were expected to support microbial specialists for both autochthonous and allochthonous DOC constituents. As resource diversity increases downstream, resource partitioning among microbial taxa may increase, allowing for lower multifunctional redundancy in streams below the treeline.
The high diversity of microbial communities makes the characterisation of functional traits and the investigation of functional redundancy challenging. To circumvent this bottleneck, we exposed microorganisms to the thymidine analogue bromodeoxyuridine (BrdU) and sequenced the BrdU-labelled DNA after immunocapturing (Yin et al., 2000); this allowed us to identify metabolically active microorganisms upon amendment with model autochthonous and allochthonous DOC, respectively (Mou et al., 2008). Extracts were amended to benthic biofilms grown in situ above, around and below the treeline in three alpine catchments, allowing us to study diversity patterns of metabolically active biofilm communities and to relate these patterns to resource specialisation.
Materials and methods
Study sites and biofilm growth
Our study sites included three headwater streams (Obertalbach=OTB, Steinriesenbach=SRB, Riesachbach=RB) in the Schladminger Tauern, Austria, ranging from 1114 to 2275 m above sea level (a.s.l.). Sparse alpine grassland and meadow dominate vegetation above the treeline (around 1600 m a.s.l), whereas spruce (Picea abies) dominates vegetation below the treeline. Streamwater chemistry was comparable across all three catchments with very low electrical conductivity (average±s.d.: 33.3±17.7 μS cm−1) and reduced pH (7.1±0.5) typical of the crystalline geology (gneissic rock) (Supplementary Table 1). DOC concentration was generally low (0.48±0.19 mg C l−1) and tended to increase in SRB and RB, but less in OTB. Nitrate (189.9±91.4 μg N-NO3 l−1) clearly increased downstream in all catchments. We grew benthic biofilms on initially sterile unglazed ceramic tiles (surface area: 2 cm2) for 8 weeks in all stream reaches in June (2011); tiles were shaded to prevent copious algal growth. Biofilms were grown across an altitudinal (and longitudinal) gradient above, around and below the treeline (Supplementary Table 1 for altitudes). Biofilm samples were transported within 3 h in sterile containers filled with 0.2 μm-filtered streamwater (cooled, in the dark) to the laboratory pending further processing.
Model autochthonous and allochthonous DOC
We produced model autochthonous and allochthonous DOC from algal cultures and spruce material, respectively. The former was produced from Nitzschia palea (Bacillariophyceae SAG 1052-3a) and Navicula pelliculosa (Bacillariophyceae SAG 1050-3) grown on WC medium (2 l) for 4 weeks. We harvested the algae by centrifugation, rinsed them two times with Milli-Q water, suspended the cells in Milli-Q water and sonicated the suspension on ice. Cells were pelleted and the supernatant sterile-filtered (0.2 μm GSWP filters; Millipore, Billerica, MA, USA). Equal concentrations of these two algal extracts were mixed. Allochthonous DOC was produced from an overnight cold water (Milli-Q) extraction of spruce material (mainly needles) freshly sampled from trees in the study catchments. The supernatant was harvested, sterile-filtered (0.2 μm) and the pH of the extract was adjusted to 7.3. DOC concentration of the extracts was determined using a Sievers 900 TOC Analyser (GE Analytical Instruments, Boulder, CO, USA). The chemical composition of the extracts was analysed after derivatisation on a gas chromatograph coupled to a mass spectrometer (LECO Pegasus 4D GCxGC-TOF-MS; Leco, Mönchengladbach, Germany).
BrdU incorporation
We used BrdU to identify active bacteria (Urbach et al., 1999; Yin et al., 2000; Mou et al., 2008). BrdU is incorporated into newly synthesised DNA upon replication and BrdU-labelled DNA can then be isolated by immunocapture (Urbach et al., 1999; Yin et al., 2000). In the laboratory, tiles were incubated in 15 ml falcon tubes (six tiles per tube) in sterile-filtered streamwater with 1 mm BrdU (Roche Diagnostics, Basel, Switzerland) and either autochthonous or allochthonous DOC was amended at a final concentration of 1 mg C l−1, corresponding to a twofold streamwater DOC concentration. An extra set of tiles was incubated in streamwater with BrdU and served as control to the DOC amendments. Treatments and controls were performed in triplicates. After 24 h incubation (12 °C, dark) and gentle horizontal shaking, biofilm microorganisms were harvested onto 0.2 μm GSWP filters after vortexing and sonication. To analyse the bulk biofilm community, we additionally harvested biofilms from six tiles per site without BrdU treatment. All filters were frozen immediately (−20 °C) pending DNA extraction.
DNA extraction and BrdU immunocapture
DNA was extracted from the biofilms using the PowerSoil DNA Extraction Kit (MoBio, Carlsbad, CA, USA). BrdU-labelled DNA was isolated according to Yin et al. (2000). In brief, 18 μl herring sperm DNA (1.25 mg ml−1; Promega, Fitchburg, WI, USA) was denatured, flash-frozen in a dry ice-ethanol bath and incubated with 2 μl BrdU antibody (Invitrogen, Carlsbad, CA, USA) for 50 min. Then, 20 μl denatured DNA extract were added and samples were incubated for 30 min, followed by the addition of 6.26 μl beads (Invitrogen) coated with an anti-BrdU antibody (Invitrogen) and incubated again. The DNA–antibody–bead complex was washed 10 times using 100 μl phosphate-buffered saline-bovine serum albumin (PBS-BSA). Finally, the labelled DNA was released from the complex by incubation with 20 μl 1.7 mm BrdU for 35 min. All incubations were carried out in the dark under continuous horizontal mixing. As methodological negative control, we included non-BrdU-labelled samples in the immunocapture procedure. BrdU-labelled DNA was purified by ethanol precipitation, where 1/10th volume of sodium acetate and 2 volumes of 100% ice-cold ethanol were added to the DNA solution and incubated (2 h, −20 °C). DNA was pelleted, the pellet washed with 70% ethanol and resuspended in sterile H2O.
PCR amplification and 454 pyrosequencing
Purified labelled DNA was further processed by PCR. The V3–V4 region of the 16S rRNA gene was amplified in a 50 μl PCR reaction containing 0.5 μmol l−1 of each primer (Thermo Scientific, Waltham, MA, USA), 0.2 mmol l−1 dNTPs (Thermo Scientific), 40 μg bovine serum albumin (Thermo Scientific), 4 mmol l−1 MgCl2 (Thermo Scientific), 2 U Taq DNA polymerase with the recommended PCR buffer (Thermo Scientific) and 4 μl DNA extract (2–4 ng DNA). Each PCR reaction included a negative control. Primers used for amplification of the 16S rRNA gene were the bacterial 341F 5′-CCTACGGGNGGCWGCAG-3′ and 805R 5′-GACTACHVGGGTATCTAATCC-3′, containing the 454 Titanium A and B adaptors, respectively (Logue et al., 2011). For each sample, two different sample-specific barcodes contained in the forward primer were used to reduce barcode-specific bias (Berry et al., 2011).
Samples were amplified using an initial denaturing step of 2 min at 94 °C, followed by 33 cycles of 30 s denaturation at 94 °C, 45 s annealing at 50 °C, 1 min elongation at 72 °C and a final elongation for 10 min at 72 °C. PCR products were run on a 1% agarose gel and purified using the Gel Extraction Kit (Qiagen, Hilden, Germany). The purified PCR products were quantified on gels (Bio-Rad, Hercules, CA, USA) and pooled equimolarly for pyrosequencing. Several samples from SRB yielded too low amounts of BrdU-labelled DNA and thus only one replicate could be used and sequenced.
Amplicons were sequenced on a GS FLX Titanium Sequencer (Centre for Genomic Research, University of Liverpool, UK). Raw output files were processed using the software package AmpliconNoiseV1.0 (Quince et al., 2011), which performed a filtering and denoising of the flowgrams. A complete linkage algorithm on a 97% identity level clustered these reads to operational taxonomic units (OTUs) (AmpliconNoiseV1.0) and taxonomic assignments were determined by using a naive Bayesian rDNA classifier (Ribosomal Database Project; Wang et al., 2007) at an 80% confidence threshold. The original pyrosequencing output files are available at the NCBI Sequence Read Archive under the accession number SRP033635.
Environmental variables and cell abundance
Environmental parameters were measured at the beginning and at the end of the 8-week growth period of biofilms. Streamwater temperature, pH and electrical conductivity were measured in situ using handheld probes (pH320, Cond340i; WTW, Weilheim, Germany). Streamwater DOC concentration was measured using a Sievers 900 TOC Analyser and concentration of N-NO3 was determined using Continuous Flow Analysis (Alliance Instruments, Salzburg, Austria). We determined microbial cell abundance in the harvested biofilms using SYTOX Green (Invitrogen) for nucleic acid staining and flow cytometry (Cell Lab Quanta SC; Beckman Coulter, Brea, CA, USA). Before staining, bacterial cells (killed and fixed in 2.5% formaldehyde) were dispersed using pyrophosphate and sonication.
Data analysis
Microbial diversity was estimated as richness of OTUs rarefied to the lowest number of reads (985) obtained for a sample using the function ‘rarefy’ in the ‘vegan’ package in R (Oksanen et al., 2013). To visualise differences in community composition, we performed non-metric multidimensional scaling on dissimilarity matrices. These dissimilarity matrices were calculated by applying the relative abundance-based Horn distance (Jost, 2007). Communities were randomly resampled to the lowest number of reads before the distance between a sample pair was calculated; each entry in the similarity matrices was then calculated as the average of 1000 such similarity values (Besemer et al., 2013). To unravel the main underlying factors explaining the distribution of samples in the ordination, that is, ‘site’ (above, around and below the treeline), ‘stream’ (OTB, SRB, RB) or ‘treatment’ (algae, spruce, control) for community structure, we performed a forward variable selection using the function ordiR2step in the ‘vegan’ package in R (Oksanen et al., 2013). This function performs stepwise model selection using a double stopping criterion based on adjusted R2 and P-values (Blanchet et al., 2008).
To identify potential specialists and generalists at family level, we used ternary plots to visualise the relative proportions of families among the different treatments and sites (above, around and below the treeline). Given that the three study streams showed similar altitudinal patterns of community composition in the non-metric multidimensional scaling, we averaged the relative abundances of the three streams to retain one plot for each site. A microbial family, of which the relative abundance was above 60%, as an arbitrary threshold, in either the autochthonous or allochthonous DOC treatment, was classified as ‘algae’ or ‘spruce’ specialist, respectively. All others were classified as generalists. We also considered the control group, as families dominating at >60% in the control treatment and hence representing taxa dominating in the absence of additional carbon substrates.
To identify potential specialists for either autochthonous or allochthonous DOC (above, around and below the treeline), we calculated indicator values for each OTU (Dufréne and Legendre, 1997) using the function indVal in the package ‘labdsv’ in R. Indicator values are calculated as the product of relative frequency and relative average abundance of a given OTU. We considered indicator species as significant for indicator values >0.3 and P-values <0.05.
We assessed metabolic functional traits using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt; Langille et al., 2013). Based on fully sequenced genomes, this algorithm predicts the metagenome, which is adjusted for 16S rRNA gene copy number to produce the KEGG (Kyoto Encyclopaedia of Genes and Genomes) classified functions of the community. Based on 140 orthologues related to carbohydrate and amino-acid metabolism, we assessed metabolic multifunctionality, and by relating multifunctionality to diversity, we estimated multifunctional redundancy (Miki et al., 2014). Metabolic functional diversity was computed as Shannon diversity, which accounts for copy number. Multifunctional redundancy was derived from a power-law function (MF=cORa) of orthologue number (i.e., MF) as a function of rarefied OTU richness. The coefficient α serves as a multifunctional redundancy index with lower values indicating larger multifunctional redundancy (Miki et al., 2014; see Supplementary Information for a detailed description).
Results
Composition of allochthonous and autochthonous DOC
The chemical composition of the model allochthonous (spruce) and autochthonous (algae) DOC that we used to amend biofilms differed clearly (Supplementary Figures 1 and 2). Compounds, which are typical for vascular plants, such as quinic acid, shikimic acid or pinitol (Yoshida et al., 1975), and also elevated concentrations of sugars, sugar acids and organic acids dominated in the spruce extract. Abundant lumichrome, glycerol and ribose, and amino acids characterised the algal extracts. Furthermore, the number of chemical compounds detected in the spruce extract (n=100) was higher compared with that in the algal extract (n=78). Moreover, the algal extract was dominated by a few compounds (e.g., glycerol), whereas the spruce extract was characterised by a more even distribution of compounds. An inverse Simpson index (i.e., a measure of dominance varying between zero and one) of 0.85 and 0.19 for the algal and spruce extract, respectively, clearly underscored this difference. These numbers translate into an elevated chemical diversity in the spruce extract.
Spatial patterns of microbial community diversity, composition and functional traits
Biofilms built up communities with cell numbers ranging from 4.2±4.0 × 106 cell cm−2 in reaches above the treeline in SRB to 22.6±5.0 × 106 cell cm−2 in reaches around the treeline in OTB (Supplementary Table 1). Microbial richness was significantly related both to altitude (Figure 1a) as a descriptor for the spatial positioning along the longitudinal gradient and to cell abundance (Figure 1b). Accounting for altitude, richness of the active (BrdU-labelled) communities was significantly lower than richness of the bulk (i.e., before BrdU incorporation) communities (analysis of covariance: F1,15=40.08, P<0.01); however, the slopes of the regression were not homogeneous (F=0.002, P=0.96). Accounting for cell counts, mean number of OTUs (log-transformed) was also significantly higher in the bulk community as compared with the BrdU-labelled community (analysis of covariance: F1,15=19.36, P<0.001), whereas the slopes were not equal (F=0.35, P=0.56). Non-metric multidimensional scaling analysis revealed a clear separation of the bulk and active communities (i.e., BrdU-labelled DNA; Figure 2). The analysis also revealed a clear downstream gradient of community composition (bulk and active) of the biofilms from above, around and below the treeline; this pattern was consistent among all three streams. Among the active communities, treatments with autochthonous and allochthonous DOC and the control clustered tightly, indicative of similar community composition among treatments. A forward variable selection identified ‘site’ (P<0.01) and ‘stream’ (P<0.01) as significant factors when all samples were included in the analysis, whereas only ‘site’ was significant (P<0.01) when only BrdU-labelled communities were considered. ‘Treatment’ was not a significant (P>0.05) factor in any of the analyses.

Microbial richness in bulk (initial; white circles) and active (BrdU-labelled, black circles) communities as a function of altitude (a) and cell counts on the tiles (b). Microbial richness was negatively related to altitude and showed a positive logarithmic relationship to microbial abundance. The error bars represent the s.d. of the replicates and streams.

Non-metric multidimensional scaling analysis of the bulk (initial) and active (BrdU-labelled) communities. Colours represent streams: green—OTB, red—SRB, blue—RB; the darkest colour represents the uppermost site (above the treeline), the lightest colour the lowest site (below the treeline). Symbols: diamond—algal treatment, circle—spruce treatment, triangles—control treatment, squares—bulk community. Bulk communities: n=9; active communities: n=63. The error bars of the active OTB and RB samples represent the s.d. of the three replicates. Although the bulk community differed clearly from the active community, the different treatments showed similar community compositions.
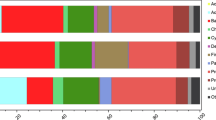
At the phylum level, Proteobacteria (average±s.d., 73±11%) and Bacteroidetes (20±8%) were most abundant across all samples, followed by Verrucomicrobia (2±2%), Actinobacteria (2±2%) and Acidobacteria (1±1%). Taxonomic composition of the active communities at the family level (50 most abundant families) changed downstream from above to around and below the treeline (Figure 3). For instance, the relative abundances of Chitinophagaceae, Rhodobacteraceae, Opitutaceae, Gemmatimonadaceae and OD1 increased downstream, whereas families such as Oxalobacteraceae, Micrococcaceae, Nocardioidaceae, Cytophagaceae, Sphingobacteriaceae, Sphingomonadaceae and Pseudomonadaceae decreased in relative abundance.

Heatmap showing the relative importance of the 50 most abundant microbial families (excluding chloroplasts and unclassified OTUs). Note that the relative abundances of each microbial family among all samples were normalised (i.e., each line sums to 1) to show the distribution of the taxa among treatments and sites, rather than the relative abundances of the taxa in the samples. This proportion is visualised using colour coding.
Ternary plots revealed some potential specialist microbial taxa but abundant generalists in the biofilms across sites and streams (Figure 4). Few specialists could be related to a predominant use of autochthonous DOC. It should be noted that these families contributed <1% to the active communities. Downstream patterns of these potential specialists were not consistent, as Leuconostocaceae (Firmicutes), for instance, preferably used autochthonous DOC around the treeline and allochthonous DOC below the treeline. On the other hand, Lachnospiraceae (Firmicutes) and Neisseriaceae (β-Proteobacteria) were more abundant in the amendments with autochthonous DOC above and around the treeline. Several families (e.g., Parachlamydiaceae and Alteromonadaceae) were identified as active in the control (i.e., no DOC amendment), as it was particularly evident in stream reaches below the treeline. Most families, however, showed comparable abundances among treatments, indicative of generalist traits. Most prominent among them were Chitinophagaceae, Oxalobacteraceae and Comamonadaceae.

Ternary plot, displaying the relative proportion of bacterial families among the different treatments and sites. Families of which more than 60% were detected in one treatment were classified specialist (indicated by the coloured lines). The size of the circles represents the relative abundance of the taxa on a logarithmic scale. Families, which constituted <0.01% of the total community (averaged over all three treatments), unclassified reads and chloroplasts were excluded from the graph. The displayed populations represent 81%, 63% and 60% of the total community from the sites above, at and below the treeline, respectively.
The indicator species analysis significantly identified 7 and 13 (P<0.05) indicator OTUs for the autochthonous and allochthonous DOC amendments among all sites, respectively (Table 1). OTUs belonging to the Oxalobacteraceae were identified as indicator species for autochthonous DOC in stream reaches above treeline, whereas OTUs belonging to the Chitinophagaceae, Spartobacteria, Pseudomonadaceae, Cytophagaceae and Alcaligenaceae were identified as indicator species for autochthonous DOC in stream reaches below treeline. Interestingly, indicator species of allochthonous DOC increased downstream, with no hit in stream reaches above treeline, one hit in reaches around and 12 hits downstream of the treeline (Table 1).
Multifunctional redundancy
Procrustes rotation revealed the coupling between functional (metabolic functional orthologues) and compositional (16S rRNA gene) similarity (Procrustes Sum of Squares 0.538, correlation 0.679, P=0.001), which exhibited congruent gradients from above to below the treeline (Supplementary Figure 3). We found significant saturating relationships between subsampled OTU richness and the Shannon diversity of orthologues for biofilms above, around and below the treeline (Figure 5), indicating multifunctional redundancy for each location. The coefficient of the power-law relationship, a measure of multifunctional redundancy, was highest for the sites above the treeline (α=0.0046±0.0004) and lowest around the treeline (α=0.0041±0.0005); downstream of the treeline the coefficient increased again (α=0.0045±0.0007); however, the confidence intervals for these estimates overlapped (Supplementary Information, MODELS). Rarefied taxon richness was higher in downstream sites (Figure 5); however, there was no obvious difference between control, algal and spruce treatments.

Metabolic orthologue accumulation curves for sites above (a), at (b) and below the treeline (c). Shown are all treatments (see legend for symbols) at the three sites (green—OTB, red—SRB, blue—RB). For each position in the landscape (a–c), the three different streams (colours) occupy different positions on the power-law function, indicating the metabolic multifunctional differences between the three locations, whereas the DOC amendments did not result in obvious changes in multifunctionality. Given are the exponents of the power-law function (dashed lines), which indicate multifunctional redundancy. Multifunctional redundancy slightly increases along the longitudinal gradient, with a peak around the treeline.
Discussion
Our study uncovers spatial patterns of biofilm diversity and community composition along longitudinal and altitudinal gradients in three Alpine streams. We found that microbial diversity increases downstream and that microbial specialists gained importance downstream of the treeline, whereas generalists dominated biofilm communities along the entire altitudinal gradient. Our findings are generally consistent with ecological theory positing relationships between resource diversity, specialisation and species richness (Kassen, 2002; Rosenfeld, 2002; Evans et al., 2005).
Microbial specialists consistently increased both at the level of OTUs and microbial families in all three study streams moving from upstream to downstream of the treeline. We propose that this pattern is associated with an increase in resource diversity along the altitudinal gradient. In fact, based on the chemical composition and diversity of the model resources used in our experiments, we would anticipate an increase in DOC diversity in reaches downstream of the treeline. Here, chemically rich terrestrial deliveries add up to in-stream sources from primary production—the dominant source of DOC upstream of the treeline where catchments are largely devoid of vascular plants. This is consistent with reports on the variation of DOC chemical composition changing with land use and soils (e.g., Wilson and Xenopoulos, 2008). It is thus intuitive to assume that elevated diversity of DOC in reaches downstream of the treeline may translate into elevated niche differentiation and specialisation, and therefore into higher diversity but lower functional redundancy (Rosenfeld, 2002; Clavel et al., 2011). The notion that the metabolism of diverse DOC may be related to microbial diversity agrees, in fact, with theory of niche partitioning among competing species (Rosenfeld, 2002; Kassen, 2002; Evans et al., 2005), whereas our findings on functional redundancy do not fully agree with our expectations.
Microorganisms assumedly specialised on autochthonous DOC did not exhibit clear gradients along the streams—and identified patterns were even conflicting between OTU and family level. This may be attributable to the fact that reduced resource diversity and variable supply, as typical for high-altitudinal streams (Battin et al., 2004; Milner et al., 2009), may prevent microorganisms from specialising on a few carbon sources. This observation is in line with recent reports on marine microorganisms showing a low degree of specialisation on carbon sources (Mou et al., 2008; Pedler et al., 2014).
It is commonly believed that environmental fluctuations support the dominance of generalists (e.g., Townsend 1989; Kassen, 2002). Environmental fluctuations in alpine streams above the treeline are particularly pronounced, often even with diurnal fluctuations in hydrology, water chemistry and resource availability (Battin et al., 2004; Milner et al., 2009). That we predominantly observed generalists in these reaches may indicate that generalist traits are an adaptive response of stream biofilms to maintain community productivity and stability (Evans et al., 2005; Matias et al., 2013).
Beside the fact that resource diversity may contribute to the observed gradient in functional traits, we also propose dispersal dynamics as an alternative or complementary mechanism. In fact, dispersal limitation is commonly believed to be higher in the headwater reaches (Besemer et al., 2013), which would induce stronger constraints on the colonisation dynamics of microbial specialists. Furthermore, the combination of elevated physical disturbance (e.g., hydrology) on microbial communities (Widder et al., 2014) upstream with the observation that postdisturbance recovery of specialists is less successful than for generalists (Clavel et al., 2011) may further contribute to the spatial patterns of functional traits we observed in this study. Finally, benthic algae which provide the bulk DOC source in streams above the treeline exude compounds, often monomeric in nature, that do not require specialised metabolic pathways and that are therefore readily available to a wide range of microbial heterotrophs (Kaplan and Bott, 1989). These combined effects may further contribute to the specialisation pattern that we observed along the study streams.
Our results suggest that functional traits were at least to some extent reflected in the taxonomy of the benthic biofilms as some taxa were overrepresented in the group classified as specialists. Indeed, half of the OTUs specialised on allochthonous DOC belonged to the Flavobacteriaceae and Chitinophagaceae (phylum Bacteroidetes; Table 1). Members of these groups have been reported to degrade complex biopolymers (Kirchman, 2002), as are generated during the degradation of plant material (Thomas et al., 2011). Interestingly, Bacteroidetes were also found to be prominent in the phyllosphere and to be more abundant on conifers than on Angiosperms (Redford et al., 2010; Thomas et al., 2011). Algal-derived DOC specialists comprised several families belonging to the Firmicutes, which is remarkable as Firmicutes constituted only a minor portion of the community (average <0.5%). This metabolically diverse group may thus comprise taxa, which are able to respond quickly to the supply of autochthonous DOC. Moreover, the Neisseriaceae (β-Proteobacteria), also classified as algal–DOC specialists, were reported to co-occur with cyanobacterial blooms (Eiler et al., 2006). The significant number of bacterial families, which had their highest relative abundance in the controls, indicates that these taxa exhibited below average activity when amended with allochthonous or autochthonous DOC. These taxa may be adapted to very low DOC concentrations (Li et al., 2012), thereby benefiting less from the amendments compared with other taxa.
We consistently found that microbial diversity decreased with altitude across the three study streams. We suggest that elevated specialisation at lower altitudes contributes to this diversity gradient. However, we do acknowledge that microbial diversity was also related to abundance and we therefore cannot exclude different growth rates, for instance, contributing to the biodiversity patterns. Several studies have predicted how microbial community composition may vary with altitude, but the relationship between microbial diversity and altitude remains debated (Bryant et al., 2008; Fierer et al., 2011). Our findings on functional traits and diversity shed new light on this relationship for stream biofilms.
Metagenome reconstruction revealed that bacterial community composition and functional similarity were coupled, and for each location along the stream, multifunctionality scaled with diversity following the suggested power-law function (Miki et al., 2014). Reaches above and below the treeline had slightly higher metabolic multifunctional redundancy compared with reaches around the treeline. Increasing resource diversity along the stream gradient may only partly explain this pattern of multifunctionality. Because the position of biofilm communities along the orthologue accumulation curves (Figure 5) did not differ between treatments, one may infer that most microorganisms in stream biofilms metabolise diverse carbon compounds from various sources despite the absence of major terrestrial deliveries above the treeline. This indicates a generally high level of multifunctional redundancy, which is indeed consistent with abundant generalists along the streams. The fact that we used Shannon diversity of orthologues rather than orthologue richness makes our estimates not directly comparable to the values reported by Miki et al. (2014).
Microbial community composition changed markedly along all three streams, whereas DOC amendments had less effect. This suggests that factors other than only resources shape community composition of the stream biofilms. Such factors may include streamwater pH and nitrate concentration, which typically increased downstream (Supplementary Table 1), and dispersal dynamics related to the hydrologic flow paths (Besemer et al., 2013). Here it is worth mentioning that the BrdU approach captures the immediate response to substrate amendments by a community that established over weeks under in situ conditions in the streams. Thus, even a slight increase in growth of one microbial population would be sufficient to induce a community shift given there is enough time. We recognise the potential caveats of BrdU incorporation to detect metabolically active microorganisms. There is evidence in fact that a certain fraction of active microbial taxa may be missed because of their intrinsic inability to incorporate BrdU (e.g., Hamasaki et al., 2007). Furthermore, despite the fact that we amended in excess, we cannot exclude that some taxa used metabolites from others. Nevertheless, BrdU incorporation seems appropriate to address resource use and specialisation in complex microbial communities (Mou et al., 2008).
We revealed altitudinal patterns of community composition, diversity, specialisation and function in benthic biofilms that were consistent among three alpine streams. Our study thus paves the way toward new mechanistic understanding of microbial diversity patterns in streams, and further supports the notion that microorganisms exhibit distributional patterns along the fluvial continuum (Besemer et al., 2013; Read et al., 2014; Savio et al., 2014) as recognised for invertebrates (Vannote et al., 1980). These findings may have broader impacts as the treeline moves upwards because of climate warming in the Alps.
Accession codes
References
Battin TJ, Kaplan LA, Findlay S, Hopkinson CS, Marti E, Packman AI et al. (2008). Biophysical controls on organic carbon fluxes in fluvial networks. Nat Geosci 1: 95–100.
Battin TJ, Wille A, Psenner R, Richter A . (2004). Large-scale environmental controls on microbial biofilms in high-alpine streams. Biogeosciences 1: 159–171.
Berry D, Karim BM, Wagner M, Loy A . (2011). Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Appl Environ Microbiol 77: 7846–7849.
Besemer K, Singer GA, Quince C, Bertuzzo E, Sloan W, Battin TJ . (2013). Headwaters are critical reservoirs of microbial diversity for fluvial networks. Proc R Soc Ser B 280: 20131760.
Blanchet FG, Legendre P, Borcard D . (2008). Forward selection of explanatory variables. Ecology 89: 2623–2632.
Bryant J, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL . (2008). Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. Proc Natl Acad Sci USA 105: 11505–11511.
Clavel J, Julliard R, Devictor V . (2011). Worldwide decline of specialist species: toward a global functional homogenization? Front Ecol Environ 9: 222–228.
Crump BC, Kling GW, Bahr M, Hobbie JE . (2003). Bacterioplankton community shifts in an Arctic lake correlate with seasonal changes in organic matter source. Appl Environ Microbiol 69: 2253–2268.
Dufréne M, Legendre P . (1997). Species assemblages and indicator species: the need for a flexible assymetrical approach. Ecol Monogr 67: 345–366.
Eiler A, Olsson JA, Bertilsson S . (2006). Diurnal variations in the auto- and heterotrophic activity of cyanobacterial phycospheres (Gloeotrichia echinulata and the identity of attached bacteria. Freshwater Biol 51: 298–311.
Evans KL, Warren PH, Gaston KJ . (2005). Species-energy relationships at the macroecological scale: a review of the mechanisms. Biol Rev 80: 1–25.
Fierer N, McCain DM, Meir P, Zimmermann M, Rapp JM, Silman MR et al. (2011). Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92: 797–804.
Gamfeldt L, Hillebrand H, Jonsson PR . (2008). Multiple functions increase the importance of biodiversity for overall ecosystem functioning. Ecology 89: 1223–1231.
Hamasaki K, Taniguchi A, Tada Y, Long RA, Azam F . (2007). Actively growing bacteria in the inland sea of Japan, identified by combined bromodeoxyuridine immunocapture and denaturing gradient gel electrophoresis. Appl Environ Microbiol 73: 2787–2798.
Hillebrand H, Matthiessen B . (2009). Biodiversity in a complex world: consolidation and progress in functional biodiversity research. Ecol Lett 12: 1405–1419.
Jaffé R, Yamashita Y, Maie N, Cooper WT, Dittmar T, Dodds WK et al. (2012). Dissolved organic matter in headwater streams: compositional variability across climatic regions of North America. Geochim Cosmochim Acta 94: 95–108.
Jost L . (2007). Partitioning diversity into independent alpha and beta components. Ecology 88: 2427–2439.
Judd KE, Crump BC, Kling GW . (2006). Variation in dissolved organic matter controls bacterial production and community composition. Ecology 87: 2068–2079.
Kaplan LA, Bott TL . (1989). Diel fluctuations in bacterial activity on streambed substrata during vernal algal blooms: effects of temperature, water chemistry, and habitat. Limnol Oceanogr 34: 718–733.
Kassen R . (2002). The experimental evolution of specialists, generalists, and the maintenance of diversity. J Evol Biol 15: 173–190.
Kirchman DL . (2002). The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39: 91–100.
Koetsier P, McArthur V, Leff L . (1997). Spatial and temporal response of stream bacteria to sources of dissolved organic carbon in a blackwater stream system. Freshwat Biol 37: 79–89.
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31: 814–821.
Lennon JT, Aanderud ZT, Lehmkuhl BK, Schoolmaster DR . (2012). Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 93: 1867–1879.
Li D, Sharp JO, Saikaly PE, Ali S, Alidina M, Alawari MS et al. (2012). Dissolved organic carbon influences microbial community composition and diversity in managed aquifer recharge systems. Appl Environ Microbiol 78: 6819–6828.
Logue JB, Langenheder S, Andersson AF, Bertilsson S, Drakare S, Lanzén A et al. (2011). Freshwater bacterioplankton richness in oligotrophic lakes depends on nutrient availability rather than on species–area relationships. ISME J 6: 1127–1136.
Matias MG, Combe M, Barbera C, Mouquet N . (2013). Ecological strategies shape the insurance potential of biodiversity. Front Microbiol 3: 432.
McArthur JV, Marzolf GR, Urban JE . (1985). Response of bacteria isolated from a pristine prairie stream to concentration and source of soluble organic carbon. Appl Environ Microbiol 49: 238–241.
Miki T, Yokokawa T, Matsui K . (2014). Biodiversity and multifunctionality in a microbial community: a novel theoretical approach to quantify functional redundancy. Proc R Soc Ser B 281: 20132498.
Milner AM, Brown LE, Hannah DM . (2009). Hydroecological response of river systems to shrinking glaciers. Hydrol Proc 23: 62–77.
Mou X, Sun S, Edwards RA, Hodson RE, Moran MA . (2008). Bacterial carbon processing by generalist species in the coastal ocean. Nature 451: 708–711.
Muller EEL, Pinel N, Laczny CC, Hoopmann MR, Narayanasamy S, Lebrun LA et al. (2014). Community-integrated omics links dominance of a microbial generalist to fine-tuned resource usage. Nat Commun 5: 5603.
Oksanen AJ, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB et al. (2013), Package ‘vegan’. In: Vegan: Community Ecology Package.
Pedler BE, Aluwihare LI, Azam F . (2014). Single bacterial strain capable of significant contribution to carbon cycling in the surface ocean. Proc Natl Acad Sci USA 111: 7202–7207.
Peter H, Ylla I, Gudasz C, Romaní AM, Sabater S, Tranvik LJ . (2011). Multifunctionality and diversity in bacterial biofilms. PLoS One 6: e23225.
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ . (2011). Removing noise from pyrosequenced amplicons. BMC Bioinform 12: 1–18.
Read DS, Gweon HS, Bowes MJ, Newbold LK, Field D, Bailey MJ et al. (2014). Catchment-scale biogeography of riverine bacterioplankton. ISME J 9: 516–526.
Redford AJ, Bowers RM, Knight R, Linhart Y, Fierer N . (2010). The ecology of the phyllosphere: geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ Microbiol 12: 2885–2893.
Rosenfeld JS . (2002). Functional redundancy in ecology and conservation. Oikos 98: 156–162.
Savio D, Sinclair L, Ijaz UZ, Stadler P, Blaschke AP, Reischer G et al. (2014). Bacterial diversity along a 2600 km river continuum. BioRxiv Available at: http://dx.doi.org/10.1101/010041.
Schoener TW . (1974). Resource partitioning in ecological communities. Science 185: 27–39.
Sun L, Perdue EM, Meyer JL, Weis J . (1997). Use of elemental composition to predict bioavailability of dissolved organic matter in a Georgia river. Limnol Oceanogr 42: 714–721.
Thomas F, Hehemann JH, Rebuffet E, Czjzek M, Michel G . (2011). Environmental and gut bacteroidetes: the food connection. Front Microbiol 2: 93.
Townsend CR . (1989). The patch dynamics concept of stream community ecology. J N Am Benth Soc 8: 36–50.
Urbach E, Vergin KL, Giovannoni SJ . (1999). Immunochemical detection and isolation of DNA from metabolically active bacteria. Appl Environ Microbiol 65: 1207–1213.
Vannote RL, Minshall GW, Cummins KW, Sedell JR, Cushing CE . (1980). The River Continuum Concept. Can J Fish Aquat Sci 37: 130–137.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Env Microbiol 73: 5261–5267.
Widder S, Besemer K, Singer GA, Ceola S, Bertuzzo E, Quince C et al. (2014). Fluvial network organization imprints on microbial co-occurrence networks. Proc Natl Acad Sci USA 111: 12799–12804.
Wilson HF, Xenopoulos MA . (2008). Effects of agricultural land use on the composition of fluvial dissolved organic matter. Nat Geosci 2: 37–41.
Yin B, Crowley D, Sparovek G, De Melo WJ, Borneman J . (2000). Bacterial functional redundancy along a soil reclamation gradient. Appl Environ Microbiol 66: 4361–4365.
Yoshida S, Tazaki K, Minamikawa T . (1975). Occurrence of shikimic and quinic acids in angiosperms. Phytochemistry 14: 195–197.
Zah R, Uehlinger U . (2001). Particulate organic matter inputs to a glacial stream ecosystem in the Swiss Alps. Freshwat Biol 46: 1597–1608.
Acknowledgements
We thank G Singer and B Eichelberger for assistance in the field and G Steniczka for assistance in the laboratory. We are most grateful for supporting discussions with C Fasching and M Bengtsson. We also thank M Striebel for providing the algal cultures. Financial support came from the Austrian Science Fund (START Y420-B17) to TJB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Wilhelm, L., Besemer, K., Fragner, L. et al. Altitudinal patterns of diversity and functional traits of metabolically active microorganisms in stream biofilms. ISME J 9, 2454–2464 (2015). https://doi.org/10.1038/ismej.2015.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.56
This article is cited by
-
Trajectories of freshwater microbial genomics and greenhouse gas saturation upon glacial retreat
Nature Communications (2023)
-
Plastic pollution fosters more microbial growth in lakes than natural organic matter
Nature Communications (2022)
-
Insect-associated bacterial communities in an alpine stream
Hydrobiologia (2020)
-
Bacterial diversity in aqueous/sludge phases within diesel fuel storage tanks
World Journal of Microbiology and Biotechnology (2020)
-
Absolute quantification of microbial taxon abundances
The ISME Journal (2017)
