
Abstract
Disentangling the mechanisms shaping bacterioplankton communities across freshwater ecosystems requires considering a hydrologic dimension that can influence both dispersal and local sorting, but how the environment and hydrology interact to shape the biogeography of freshwater bacterioplankton over large spatial scales remains unexplored. Using Illumina sequencing of the 16S ribosomal RNA gene, we investigate the large-scale spatial patterns of bacterioplankton across 386 freshwater systems from seven distinct regions in boreal Québec. We show that both hydrology and local water chemistry (mostly pH) interact to shape a sequential structuring of communities from highly diverse assemblages in headwater streams toward larger rivers and lakes dominated by fewer taxa. Increases in water residence time along the hydrologic continuum were accompanied by major losses of bacterial richness and by an increased differentiation of communities driven by local conditions (pH and other related variables). This suggests that hydrology and network position modulate the relative role of environmental sorting and mass effects on community assembly by determining both the time frame for bacterial growth and the composition of the immigrant pool. The apparent low dispersal limitation (that is, the lack of influence of geographic distance on the spatial patterns observed at the taxonomic resolution used) suggests that these boreal bacterioplankton communities derive from a shared bacterial pool that enters the networks through the smallest streams, largely dominated by mass effects, and that is increasingly subjected to local sorting of species during transit along the hydrologic continuum.
Similar content being viewed by others

Introduction
The widespread use of high-throughput sequencing approaches in microbial ecology studies is resulting in unprecedented insight into the structure of prokaryotic communities. We now know that the taxonomic composition of bacterial communities differs greatly between types of ecosystems (Pommier et al., 2006; Lozupone and Knight, 2007; Barberán and Casamayor, 2010) and within ecosystems at different temporal and spatial scales (Gilbert et al., 2012; Jones et al., 2012; Shade et al., 2013; Cottrell et al., 2015). In general, these differences are driven by two major mechanisms: (1) dispersal of microorganisms among communities, which determines the bacteria that can be transported and potentially colonize a given ecosystem, and (2) selection of species (that is, species sorting) from within this immigrant pool on the basis of their environmental preferences and local conditions (Martiny et al., 2006; Hanson et al., 2012; Lindström and Langenheder, 2012; Székely et al., 2012).
Although there is ample evidence supporting a major role of environmental sorting in shaping bacterial community structure across many different habitats types (Van der Gucht et al., 2007; Logue and Lindström, 2010; Langenheder and Kely, 2011; Logares et al., 2013; Adams et al., 2014; Souffreau et al., 2015), in freshwater ecosystems the influence of mass effects (that is, the structuring of bacterial communities by massive advection of bacteria from other systems) can be particularly important because the flow of water through the landscape often results in strong connectivity and intense exchange of microbes among local communities (Lindström et al., 2006; Crump et al., 2007, 2012; Nelson et al., 2009; Ruiz-González et al., 2015b). It is thus likely that both the environmental and hydrological conditions interact to shape the patterns that we observe in freshwater bacterioplankton communities, but how this interaction influences the biogeography of bacterioplankton over large spatial scales remains largely unexplored.
At the catchment scale, the movement of water within the network has been shown to have a key role in modulating the relative importance of species sorting and mass effects in freshwater bacterioplankton community assembly: whereas the rate of water (and thus bacterial) inputs relative to the ecosystem volume determines the intensity of the mass effects, the resulting water residence time (WRT) in turn influences the extent of species sorting by local conditions by bracketing the time available for bacterial growth (Crump et al., 2004, 2007; Lindström and Bergström, 2004; Lindström et al., 2006). In addition, because water and bacteria move directionally, most ecosystems act both as recipients and sources of bacteria, and the immigrant pool arriving to a given ecosystem will also vary depending on its position along the hydrologic continuum (Nelson et al., 2009). The local bacterioplankton community composition within a given aquatic network will thus be determined by the hydrology, the position of the system in the network, and the local environmental conditions, and consequently, the relative importance of the community assembly processes will likely vary along the hydrologic continuum.
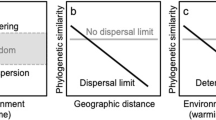
Beyond individual catchments and over broader spatial scales, however, bacterial taxonomic composition may further differ between communities as a result of increases in environmental ranges relative to the local conditions, but also due to dispersal limitation among different geographic regions (for example, Lear et al., 2013; Liu et al., 2015), which will determine the composition of the regional species pool. Thus, the large-scale spatial patterns in composition of freshwater bacterioplankton communities likely result from combinations of these local and regional processes, as conceptualized in Figure 1. For example, we may detect differences in the composition of bacterial communities inhabiting ecosystems with long WRTs (for example, lakes) either because they are located in areas that do not share the same regional bacterial pool because of low dispersal (Figure 1a), or because their environmental differences may have selected different taxa from a shared pool of bacteria (Figure 1b). Similarly, communities subjected to strong mass effects, (for example, inhabiting headwater streams with short residence times) may show differences in composition if influenced by distinct bacterial pools (Figure 1a), but may be compositionally similar if derived from a common immigrant pool (Figure 1b), regardless of their local environmental conditions. An important corollary of this conceptual framework is that disentangling the major processes underlying the spatial patterns in bacterial community composition requires incorporating the full range of variation in terms of ecosystem position, hydrologic regime and environmental conditions within and across networks located in distinct regions.

Schematic representation of potential large-scale spatial patterns in bacterioplankton taxonomic composition across freshwater networks, where dots represent individual bacterioplankton communities in an ordination space, based on their compositional dissimilarity, and colors indicate the different catchments or geographic regions to which they belong. Owing to the movement of the water in the landscape, the composition of local bacterioplankton communities within a single catchment or region will differ depending on their position along the hydrologic continuum (horizontal axis), as they will be differentially affected by hydrology that regulates the mass effects versus environmental sorting ratio (see Introduction section for further explanation), represented here by the light to dark color gradient. As a result, the compositional variation because of local conditions (vertical axis) will likely increase along the hydrologic continuum because of a gradual intensification of the local sorting of species with increasing WRT. Over broader spatial scales, the increase in environmental differences will likely result in stronger environmentally driven dissimilarities between communities (that is, along the vertical axis), but the overall large-scale spatial patterns may further differ depending on the degree of dispersal of microbes between regions or catchments: For example, in a case of low dispersal between regions (a), a regional structuring of the communities may be detected even in systems with little or no environmental sorting (that is, headwater streams) because the regional bacterial pools are different due to dispersal limitation. In contrast, under a scenario of high dispersal between regions (b), a common bacterial pool will lead to little environmentally driven compositional differences in the headwaters but to a increasing differentiation of communities toward downstream systems (that is, lakes) because of stronger local sorting of species. From this perspective, whereas communities located at the headwaters of the aquatic continuum will mostly reflect the nature of the regional bacterial pools because of strong mass effects, communities located further downstream in the network will be the result of species sorting by the local aquatic conditions. The interpretation of the factors and mechanisms underlying the observed spatial patterns in bacterial community composition will thus depend on the portion of this complex space that is considered (dotted areas, scenarios 1, 2 and 3). For example, studies based on a narrow spatial scale, as depicted in scenario 1, may conclude that hydrology is the main driver of changes in bacterioplankton community composition. In contrast, studies that cover broader environmental gradients but are limited to certain portions of the hydrologic continuum (scenarios 2 and 3) may conclude that either dispersal limitation (2) or local sorting (3) shape local community assembly. We argue that a cross-regional, whole network approach is necessary to disentangle the influence of the mechanisms and factors that are actually influencing the local assembly of bacterioplankton communities in complex freshwater networks.
Most studies to date, however, have covered only limited portions of this multidimensional space. For example, the literature abounds in studies focusing on a single type of aquatic ecosystem (for example, only lakes, only rivers; Figure 1—scenarios 2 and 3) varying across spatial scales and environmental gradients (Yannarell and Triplett, 2005; Fierer et al., 2007; Van der Gucht et al., 2007; Heino et al., 2014; Ren et al., 2015). These studies typically conclude that local conditions have a major role in shaping local community composition through species sorting. On the other hand, studies that have included multiple ecosystems (for example, streams, rivers, lakes; Figure 1—scenario 1), which may cover wide ranges of network positioning and hydrologic conditions but that are restricted to small spatial scales (for example, a single catchment, Lindström et al., 2006; Crump et al., 2007, 2012; Nelson et al., 2009; Read et al., 2015), often conclude that hydrology and the associated mass effects are major drivers of community assembly. The framework presented in Figure 1 suggests that the ambiguous and sometimes contradictory conclusions found in the literature concerning the main drivers of the spatial variability in aquatic bacterial community composition are neither incorrect nor mutually exclusive, but rather represent fragments of a more complex biogeographic reality. Understanding the mechanisms underlying this large-scale biogeography thus requires a cross-regional, whole network perspective that has seldom, if ever, been undertaken.
Here we explore the large-scale spatial patterns in freshwater bacterioplankton community composition across the boreal biome of Québec, and we infer its main underlying drivers and mechanisms by exploring the variability in bacterial taxonomic structure over broad ranges in hydrologic and environmental conditions, network position, and geographic distance. We have characterized the taxonomic composition of 386 bacterioplankton communities inhabiting freshwater ecosystems positioned along the entire aquatic continuum, ranging from the smallest headwater streams to the largest rivers and lakes, which belong to seven different geographic regions that span large gradients in environmental, climate and landscape properties. This sampling design allowed us to understand how local environmental and hydrologic conditions, network position, and dispersal-related regional differences interact to shape bacterial community composition across these complex boreal aquatic networks.
Materials and methods
Study sites and sampling design
Water samples were collected from 198 lakes and 188 rivers across seven boreal regions of Québec (Canada) between 2009 and 2013. The regions sampled (Laurentides, Abitibi, James Bay, Saguenay, Chibougamau, Schefferville, La Côte-Nord; Figure 2a) cover a total area of ca 900 000 km2 (44–56°N, 64–80°W) and span large gradients in terms of climate, limnological properties, land cover and geomorphology typical of the boreal zone (for details, see Lapierre et al., 2015; Rasilo et al., 2015; Ruiz-González et al., 2015a). The studied sites covered the entire range in lake area (0.002–4345 km2) and river order (Strahler order 0–8), and were sampled once during the summer (July/August). All the sites in each region were sampled within the same year. Samples were taken at a depth of 0.5 m at the deepest measured spot of lakes and near the shore in rivers. Temperature, dissolved oxygen, pH and conductivity were measured in situ with a YSI probe. Samples were filtered in situ through 0.45 μm and stored in acid-washed glass vials for dissolved organic carbon (DOC) and optical analyses, or kept in the dark in acid-rinsed bottles for further processing.

(a) Distribution of sampling sites across the seven sampled regions in Northern Québec (Canada). (b) Principal component analysis (PCA) of the sites based on the measured environmental and geographic parameters. Different regions are indicated by different colors and symbols indicate rivers (triangle) or lakes (circles). The two first axes explain 44.5% of the variance. Chl a, chlorophyll a; Cond, conductivity; DIC, dissolved inorganic carbon; %C2, percentage of fluorescent component C2 (humic-like DOM); %C5, percentage of fluorescent component C5 (freshly produced labile DOM); Elevation, mean catchment elevation; Mean T, mean annual temperature; NPP, net primary productivity; Precipitation, mean annual precipitation; Runoff, mean annual runoff; %Forest/%Shrubland/%Wetland, %forest/shrubland/wetland covered area in the catchment; Temp, water temperature; TP;TN, total phosphorus and nitrogen.
Chlorophyll, chemical and optical analyses
Chlorophyll a concentration was determined spectrophotochemically in hot ethanol extracts (90%). DOC concentration was measured on an OI 1010, OI Analytical (College Station, TX, USA). Total phosphorus (TP) and nitrogen (TN) concentrations were analyzed after persulfate and alkaline persulfate digestions, respectively, following standard methods (see Rasilo et al., 2015).
The optical properties of DOC were measured as indices of its composition. Colored dissolved organic matter (cDOM) was quantified as the absorbance at 440nm using an Ultrospec 3100 spectrophotometer (Biochrom, Cambridge, UK). DOC composition was described on the basis of fluorescence absorption/emission spectra (EEMS), measured in a Shimadzu RF5301 PC spectrofluorophotometer (Biochrom) across excitation/emission wavelengths of 275–450 and 280–600 nm, respectively. Six main fluorescence components related either with refractory-humic (C1–C3) or with biolabile, freshly produced DOM (C4–C6) material were recovered from the EEMS using parallel factor analysis (PARAFAC, for details, see Lapierre and del Giorgio, 2014). The percent contribution of each component was calculated relative to the total fluorescence of the six PARAFAC components.
Geographic analyses
Lake areas, river length and order, catchment areas, the area covered by water in the catchment, and elevation of the sampled sites were derived using the ArcMap 10 and ArcGIS V10 software (ESRI Inc., Redland, CA, USA) applied on the digital elevation models derived from (1:50 000) maps. For each catchment area, we averaged catchment slope based on digital elevation models, as well as various land cover properties obtained from Geobase (2009). Mean annual temperature, precipitation and runoff were extracted at each site location from a long-term climate database (WordClim, Hijmans et al., 2005). The WRT of lakes was estimated from lake volume (as mean depth x lake area), catchment area, and mean annual regional runoff. River WRT was calculated based on the measured water velocity at the sampling point and the total upstream distance.
Water isotopes
Samples were collected to determine 18O and 2H isotopes by laser spectroscopic analysis of liquid water technique (LGR DT-100 Liquid Water Stable Isotope Analyzer, Los Gatos Research Inc., Mountain View, CA, USA). We used water isotopic composition to calculate the deuterium excess (d-excess), as:

which provides an index of the degree of evaporation relative to precipitation, and therefore, the relative proximity to groundwater sources. As headwater systems are strongly influenced by soil groundwater, the water carries an isotopic signature that closely resembles that of precipitation (high d-excess). Given that temperature increases and d-excess decreases as water transits through the network and is subjected to warming and evaporation (Gibson et al., 2002; Turner et al., 2014), here we use water temperature and d-excess as proxies of the groundwater-mediated connectivity with the surrounding catchment.
Bacterial community composition
In all, 300–500 ml water samples were filtered onto 0.22 μm pore-size filters, and genomic DNA was extracted from the filters using the MoBio PowerWater DNA extraction kit (Mo Bio, Carlsbad, CA, USA) following the manufacturer’s protocol. Libraries of the V3–V4 region of the 16S ribosomal RNA were made using the primers 515F and 806R, and sequenced on an Illumina MiSeq2000 (Illumina, San Diego, CA, USA) following a paired-end approach (Caporaso et al., 2012). Paired-end reads were assembled with FLASH (Magoč and Salzberg, 2011) and sequences between 250 and 290 bp were used for downstream analyses in QIIME to remove primers and low-quality, archaeal and chloroplast reads (Caporaso et al., 2010). After the detection and removal of chimeric sequences with ChimeraSlayer (Haas et al., 2011), quality sequences were aligned with Mothur aligner (Schloss et al., 2009) and clustered into operational taxonomic units (OTUs, ⩾97% similarity) using UCLUST (Edgar, 2010). In order to assign the taxonomy to the representative sequences, we used the RDP classifier and the Ribosomal Database Project taxonomy (Wang et al., 2007). We discarded all OTUs that were represented in <10 samples and/or by <10 sequences. To enable comparisons between samples, the OTU table was randomly subsampled to ensure an equal number of sequences per sample, based on the sample with the least number of reads (50 323 sequences). The sequences have been deposited in the European Nucleotide Archive, under accession number PRJEB11530.
Statistical analyses
We used nonmetric multidimensional scaling (Vegan metaNMDS, Oksanen et al., 2015) with Bray–Curtis distances to ordinate the samples based on their dissimilarity in community composition, and the Vegan envfit function (Oksanen et al., 2015) to fit environmental vectors onto the ordination space. Differences in environmental and taxonomic composition among categories (that is, regions, ecosystems) were tested using Permanova with 999 permutations (Anderson, 2006; Anderson, 2011). To estimate the relative contribution of environmental and hydrological factors in explaining the variation of the large-scale patterns of bacterial community composition, we performed a variation partitioning analysis using the non-metric multidimensional scaling (NMDS) scores as the response variables. We grouped the variables as environmental or hydrological factors, which, after a forward selection procedure, were used as the explanatory groups of variables (Borcard et al., 2011). All the analyses were made using the R 3.0.0 software (R Core Team, 2013).
Results
Large-scale patterns in bacterial community composition across boreal aquatic ecosystems
The studied lakes and rivers segregated into clearly differentiated groups of sites based on their environmental, climatic and landscape properties (Figures 2a and b). These groups roughly corresponded to the different geographic regions (PermanovaBY_REGION R2=0.43, P<0.01), although systems located in Laurentians, Côte-Nord and Saguenay tended to overlap. Sites were segregated among these regional groups based on differences in water chemistry (pH, DOC, dissolved inorganic carbon and cDOM) and trophic status (TP, TN and chlorophyll a), and lakes and rivers from a given region tended to cluster together. Briefly, the sites varied along a gradient from alkaline and more productive waters in Abitibi and Bay-James to acidic and less productive waters in Saguenay and Côte-Nord. These environmental gradients were also related with changes in mean annual temperature and regional net primary productivity (as illustrated by the PC1 axis), as well as mean annual precipitation and runoff (PC2).
This environmental segregation of sites was not as clearly reflected in terms of the composition of bacterial communities, and the NMDS analysis showed much smaller differences between the geographical regions on the basis of bacterial composition (Figure 3a, PermanovaBY_REGION R2=0.10, P<0.01), and a significant overlap in composition between sites from different regions (Figure 3a). River and lake communities, in contrast, showed a clearer segregation, although there was some degree of overlap between them (Figure 3, PermanovaBY_ECOSYSTEM R2=0.11, P<0.01), as the largest rivers (Strahler order >4) clustered together with lakes (details not shown). Interestingly, river communities were much more dispersed along the NMDS1 axis than lake communities (Figure 3).

NMDS ordination of bacterioplankton communities based on the Bray–Curtis dissimilarity of community composition (stress =0.13). Shape indicate rivers (triangles) or lakes (circles), and sites are colored according to geographical region (a) and water pH (b), which was the variable that best fitted the ordination space (R2=0.61, see Table 1). The arrows indicate the direction at which the environmental and hydrological vectors fit the best (using envfit function) onto the NMDS ordination space. The size of the arrow is proportional to the strength of the correlation of each variable. AWC, area of water in the catchment; CA, catchment area; Cond, conductivity; DIC, dissolved inorganic carbon; %C2, percentage of fluorescent component C2; %C5, percentage of fluorescent component C5 (freshly produced labile DOM); T, water temperature.
Drivers of bacterial community composition across freshwater ecosystems
In order to identify the drivers explaining the spatial patterns in community composition depicted by the NMDS in Figure 3, we fit the measured environmental and landscape variables onto the ordination. pH, WRT and water temperature were the variables most strongly correlated to the two NMDS axes (Figure 3, Table 1). NMDS1 was strongly correlated to hydrologic and landscape variables such as water temperature, WRT, d-excess, catchment area and the area of water in the catchment. This NMDS axis also represented to some extent network positioning, with scores declining with increasing river order along the hydrologic continuum, and also with increasing distance to the headwaters, thus separating small headwater streams from rivers, and lumping together the largest rivers with lakes (details not shown). NMDS2 was mostly related to water chemistry, and particularly to pH (but also to cDOM, dissolved inorganic carbon and specific DOM fluorescence components; Figure 3, Table 1).
The geographic location (latitude and longitude) was not significantly related to either of the NMDS axes, and there was no significant relationship between the spatial distance and the taxonomic dissimilarity between sites (Partial Mantel R=0.06 P=0.1; Figure 4a), thus indicating that the observed regional segregation described above (Figure 3a) was not primarily driven by dispersal limitation. Rather, the community dissimilarity was significantly correlated to both the environmental and hydrologic distances between pairs of sites (Figures 4b and c, respectively). Environmental and hydrologic distances were only weakly correlated to each other (details not shown), such that any given range of hydrologic conditions was associated to the entire range in environmental variability, suggesting that our sampling scheme captured the full range in both dimensions that exists across the boreal biome.

Relationship between geographic (a), environmental (b) and hydrological (c) distances and pairwise bacterial community dissimilarity (Bray–Curtis). pH and WRT (log10-WRT) were used as proxies of environmental and hydrological distances, respectively. Mantel correlations (R) an the probabilities are provided for each case.
Changes in taxonomic diversity and composition along the hydrologic continuum
In order to further understand the observed changes in community composition along the hydrologic continuum, we divided the sampled sites into seven groups (G1–G7) that were equally spaced along the NMDS1 axis. These groups were associated to different average network position and WRT within the hydrologic continuum: most lakes were contained within groups G1 and G2, the smallest headwater streams were mostly included in G7 and the remainder of the rivers and some lakes were distributed among the other groups. For each of the seven groups, we calculated the average OTU richness, average Pielou's evenness index and the percentage of sequences within the major bacterial Phyla. This analysis revealed that sites with the highest scores on the NMDS1 axis (G7, smallest streams) showed the highest average OTU richness (Figure 5a) and evenness (Figure 5b), and that there was a striking decline in both indices toward sites with the lowest average NMDS1 scores (G1, larger rivers and lakes; Figures 5a and b): on average, 6000 OTUs per community were lost between these two groups of sites, and the evenness decreased from 0.86 to 0.66. The taxonomic composition also changed gradually along this NMDS1 axis, resulting in highly differentiated communities at the extremes of this hydrologic continuum even at the phylum level (Figure 5c). Overall, although Actinobacteria, Bacteroidetes and Cyanobacteria increased toward larger rivers and lakes, Betaproteobacteria increased toward larger rivers but decreased again in lakes. The remaining classes of Proteobacteria and dominant soil groups such as Acidobacteria systematically decreased from the smallest streams to lakes.

Changes in OTU richness (a), Pielou’s evenness (b) and taxonomic composition (at the phylum level, c) for sites grouped according to their position on the NMDS1 axis (G1 to G7, n=174, 74, 37, 18, 26, 39, 18), which mostly represents an hydrologic continuum from the smallest headwater streams (G7) to larger rivers and lakes (G1, for details see Results section). Dots are means and error bars represent the standard error of the values for the sites within each NMDS1 group. Colors indicate different phyla (and different classes within the Phylum Proteobacteria, indicated by the dashed line) and the heights of the bars represent the percentage of sequences associated to each taxonomic rank relative to the total number of sequences within each group of sites.
Shifts in the relative influence of the environment versus hydrology on bacterial community composition
The pronounced loss of OTUs downstream the hydrologic continuum, together with the decline in taxonomic evenness associated to the dominance of certain bacterial groups (Figure 5c), suggest that the relative importance of mass effects and environmental sorting of species may change along the hydrologic continuum. To test this hypothesis, we calculated the correlation between taxonomic and environmental dissimilarity matrices for each of the seven groups of sites along the NMDS1 axis (G1 to G7, described above; Figure 6a). We used differences in pH as a proxy of environmental distance because it was the variable most strongly correlated to changes in taxonomic composition along the NMDS2 axis (Figure 3, Table 1). The correlation between the environmental and the compositional dissimilarities between sites were weakest in the groups with the shortest average WRTs and highest position within the network (that is, smallest headwater streams, G7, Mantel R=0.25), and increased gradually toward larger rivers and lakes, where changes in pH explained a much larger portion of the variability in taxonomic composition (G1, Mantel R=0.49; Figure 6a).

(a) Variation in the R coefficient of the Mantel correlations between the taxonomic and the environmental (pH) dissimilarity matrices for sites grouped according to their position on the NMDS1 axis (G1 to G7). Note that G1 includes most lakes and the largest rivers, and G7 (G1 to G7, n=174, 74, 37, 18, 26, 39, 18) contains mostly small headwater streams (for details see Results section). All the correlations were significant (P<0.01). (b) Changes in bacterial community composition along the NMDS1 axis as a function of WRT. The NMDS1 scores of the sites were binned into 16 equal groups (n=20) based on ranked WRT. The dots are the average NMDS1 scores within each bin and represent the mean position of the sites within a given bin on the NMDS1 axis. Error bars are the standard errors for either WRT and scores within each bin. Note that increases in WRT result in gradual changes in taxonomic composition along the NMDS1 axis until a WRT of 10 days is reached, above which increases in WRT do not translate into further changes in taxonomic composition along the NMDS1 axis. (c) Percentages of variation in the NMDS ordination space explained by the environment and hydrology, for sites with WRTs above and below 10 days (WRT>10 and WRT<10, respectively). Non-explained and shared variation (environment+hydrology) are also shown. The ‘environment’ category included the variables DIC, dissolved inorganic carbon; %C2, percentage of fluorescent component C2 (humic-like DOM); %C5, percentage of fluorescent component C5 (freshly produced labile DOM) and conductivity. T, water temperature; CA, catchment area; AWC, area of water in the catchment; WRT, and d-excess were included within the ‘hydrology’ category.
To explore whether there are thresholds along the hydrologic continuum that modulate the relative importance of hydrology and environment sorting, we analyzed the changes in NMDS1 scores as a function of WRT (Figure 6b), and found that above a 10-day threshold, an increase in WRT did not trigger further changes in community composition along the NMDS1 axis. Thus, in order to compare the relative influence of the environmental and hydrologic conditions on the dissimilarity between communities within the NMDS ordination space, we performed a variation partitioning analysis dividing the data set into two groups of sites based on whether their estimated WRT was >10 or <10 days (Figure 6c). This exercise clearly shows that environmental variables explained most of the variation (65%) in bacterial taxonomic composition at those sites with high WRT (>10 days), whereas hydrological variables explained a larger share of the variation (33%) than the environment (14%) in sites with shorter WRT (Figure 6c).
Discussion
Our results show that environmental sorting is a major driver of bacterial community assembly across the boreal biome, but further indicate that hydrology and network position, and the associated mass effects, also have a fundamental role in shaping bacterioplankton communities in complex aquatic networks. Although previous studies had identified these two processes as major mechanisms of freshwater bacterial community assembly (Crump et al., 2007; Fierer et al., 2007; Nelson et al., 2009; Ren et al., 2015; Souffreau et al., 2015), no study to date had explored how they interact to shape the large-scale biogeography of freshwater bacterioplankton communities. Assessing this interaction necessarily requires expanding the conceptual framework proposed by Martiny et al. (2006) to incorporate, in addition to the environmental gradients and geographic distance (as a proxy for dispersal limitation), a hydrologic dimension that controls the relative importance of the mass effects on community assembly, as we conceptualize in Figure 1.
Our results show no significant relationship between geographic distance and the taxonomic dissimilarity among the studied bacterioplankton communities (Figure 4a), suggesting that dispersal limitation between aquatic bacterial communities is negligible across boreal Québec, at least at the taxonomic resolution of our analysis. Rather, there was an overall positive relationship between environmental and taxonomic distances (Figure 4b), and communities appeared to be strongly structured by local water chemistry (mostly pH; Figure 3b), pointing to a major role of environmental sorting in structuring the large-scale biogeography of boreal freshwater bacterioplankton. We should note that although pH emerged as the most important environmental variable, in agreement with previous studies (Fierer et al., 2007; Ren et al., 2015; Ruiz-González et al., 2015b), it is likely that besides directly influencing cell physiology, pH is also an integrator of other variables of relevance for bacteria (Fierer et al., 2007). In particular, we found that changes in water pH were strongly correlated to shifts in features linked to the amount and quality of DOM (details not shown), also known to shape bacterial community structure and functioning (Fujii et al., 2012; Ruiz-González et al., 2015a; Wilhelm et al., 2015).
Despite this strong effect of pH, the fact that in environmentally similar sites we observed the entire range of community dissimilarity (Figure 4b) suggests that mechanisms other than environmental sorting are involved in shaping the large-scale biogeography of boreal freshwater bacterioplankton communities. In this regard, our results show that WRT alone explains as much variability in composition as pH (Figures 4b and c), and that bacterial communities change largely along a hydrologic continuum from headwater streams toward large rivers and lakes, following variations in WRT and network position (Figures 3 and 5).
We hypothesize that this directional structuring is caused by a gradient of decreasing mass effects and increasing environmental sorting toward downstream ecosystems as conceptualized in Figure 1. In a companion study where we characterized the soil communities surrounding aquatic systems within a single boreal region (Ruiz-González et al., 2015b), we show that whereas headwater streams are strongly dominated by bacteria washed from soils, which gradually disappear along the hydrologic continuum, downstream ecosystems are numerically dominated by a few of these soil-derived taxa that have the potential to grow in the aquatic environment. The high OTU richness found in our headwater streams across the boreal biome (Figure 5a) thus likely results from the massive immigration of terrestrial bacteria (Besemer et al., 2013; Savio et al., 2015). Moreover, we found that variations in d-excess (our proxy of groundwater-mediated connectivity with land) also explained changes in community composition (Figure 3b), supporting that these mass effects depend on the degree of connectivity to the source of immigrants (that is, the soils). In turn, the observed loss of bacterial taxa toward downstream ecosystems was accompanied (and probably caused) by gradual increases in the relative abundances of some common freshwater bacterial groups (sensu Newton et al., 2011, see examples in Supplementary Figure S1), suggesting that environmental sorting progressively increases along the hydrologic continuum. In support of this idea, and in accordance to our conceptual framework (Figure 1), we observed that the spatial patterns of the bacterial assemblages inhabiting systems with longer WRT were much more strongly influenced by differences in local aquatic conditions than those from communities inhabiting headwater streams (Figure 6a).
Interestingly, the relative influence of hydrology and the associated mass effects on community composition was not continuous: we observed a clear threshold of WRT of around 10 days, beyond which increases in WRT did not result in further hydrologically driven changes in taxonomic composition (Figure 6b). Our variance partitioning analysis supports this hypothesis, demonstrating that hydrology explains a much larger fraction of the variance in taxonomic composition in systems below this WRT threshold, whereas environmental factors were responsible for most of the variability in ecosystems with longer WRT (Figure 6c). In the latter systems, pH seemed to strongly influence OTU richness, which peaked at pH levels close to neutrality (Supplementary Figure S2a), a pattern that has been observed before (Fierer and Jackson, 2006; Ren et al., 2015). This suggests that extreme environmental conditions may impose a stronger filter for bacteria, and that neutral pH may favor the co-existence of taxa with overlapping ranges of tolerance. This relationship was lost, however, at sites with WRT>10 days (Supplementary Figure S2b), indicating that intense mass effects may overwhelm species sorting even under extreme environmental conditions. Remarkably, we identified a similar WRT within a regional aquatic network (Ruiz-González et al., 2015b), beyond which there was no further selection or growth of soil-derived bacteria along the hydrologic continuum. This threshold is likely determined by the balance between the potential for in situ growth of the resident bacterial communities relative to the rate of dilution by allochthonous bacteria (for example, Crump et al., 2007), but this issue needs further investigation.
Considering that the headwater systems are largely subjected to mass effects, we argue that the bacterial composition in these communities must reflect the nature of the terrestrial source communities that feed these networks. The question then becomes whether there are distinct pools of immigrant bacteria separated by dispersal barriers, or whether all aquatic communities derive from a common pool of taxa, at least at the scale of our study. The lack of a relationship between community dissimilarity and geographic distance discussed above suggests that the boreal biome represents a single bacterial province sensu Martiny et al. (2006), and thus that there is low dispersal limitation of taxa between regions. We are aware, however, that there could be dispersal limitation at finer taxonomic resolution, although in a previous study we have shown that the level of taxonomic aggregation used here (97%) resulted in the clearest spatial and temporal patterns in community composition from boreal and temperate lakes (Cottrell et al., 2015).
In our conceptual framework, a common bacterial pool across all regions should lead to small differences in community structure between systems that are overwhelmingly dominated by mass effects (Figure 1b), a scenario that is not supported by our results. Rather, we observed that the taxonomic differences between systems with the lowest WRT were as large as those between lake communities (Figure 3), a pattern that resembles that of Figure 1a, where dispersal limitation has a major role. As our data suggest that this is not the case, we hypothesize that the observed differences in taxonomic composition between small headwater streams reflect a sorting that occurs in the soils before the loading of terrestrial bacteria to the aquatic network. This may indicate the occurrence of a shared pool of taxa that undergoes re-arrangements driven by local landscape conditions. Thus, although there is evidence that even in headwater streams there is already some degree of aquatic sorting of terrestrially derived taxa (Ruiz-González et al., 2015b), we argue that the communities from headwater aquatic systems that are subjected to strong mass effects must largely reflect differences in the composition of the surrounding soil assemblages. In this regard, the regional signature observed in headwater streams (Figure 3a) could easily be interpreted as evidence of dispersal limitation (Figure 1a). However, the fact that low-pH headwater streams from Schefferville clustered together with equally acidic sites in Côte-Nord (Figures 3a and b), for example, suggests that the observed patterns are the result of a regional structuring of pH (and likely other covarying factors), and therefore, of pH-driven differences in soil bacterial taxonomic composition (Fierer and Jackson, 2006). This could be considered analogous to historical or legacy effects (Martiny et al., 2006) because it occurs outside the aquatic network and before the aquatic sorting, but is nevertheless based on contemporary environmental ranges and not necessarily on past events linked to dispersal limitation.
Once in the aquatic network, these migrant communities undergo further selection by local conditions as they move downstream, and especially in systems with long WRTs (Crump et al., 2012; Ruiz-González et al., 2015b), such that the composition of these communities will be mainly a reflection of the local aquatic environment (for example, Yannarell and Triplett, 2005; Fujii et al., 2012; Ren et al., 2015; Souffreau et al., 2015). This process renders lake or large river communities clearly distinct from those of headwater streams (Figures 3 and 5) within a given region, even when they may share similar ranges in key environmental factors. We recognize that our results provide a midsummer snap-shot and do not incorporate the potential temporal variability within communities, but when the taxonomic composition was analyzed for a subset of rivers and lakes sampled on three occasions (spring, summer and fall, n=331, data not shown), the same pattern was observed: irrespective of the season, aquatic communities were structured along an hydrologic and a pH gradient, highlighting the consistency and strength of the spatial patterns presented here.
Overall, we show that hydrology and network position interact with environmental gradients to shape the spatial patterns of bacterioplankton community composition across boreal freshwater networks. Our results indicate that hydrology and network position modulate the relative role of environmental sorting and mass effects on community assembly along the aquatic continuum, likely by defining both the time frame for bacterial growth and the composition of the immigrant pool. We found no evidence of dispersal limitation even at the large spatial scale of our study, but we observed a certain regional signature likely caused by the spatial structuring of environmental conditions in the landscape. We suggest that this landscape structuring influences the headwater communities through a pre-sorting of the immigrant pool of bacteria, and the communities within systems with longer WRT by determining the local aquatic conditions that bacteria will face. Despite that these two processes can generate similar spatial patterns, they represent fundamentally different mechanisms, highlighting the fact that interpretations based on partial fragments of this complex biogeographic reality may lead to a biased understanding of the factors shaping bacterioplankton communities over large spatial scales.
References
Adams HE, Crump BC, Kling GW . (2014). Metacommunity dynamics of bacteria in an arctic lake: the impact of species sorting and mass effects on bacterial production and biogeography. Front Microbiol 5: 82.
Anderson MJ . (2011). A new method for non-parametric multivariate analysis of variance. Austral Ecol 26: 32–46.
Anderson MJ . (2006). Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62: 245–253.
Barberán A, Casamayor EO . (2010). Global phylogenetic community structure and β-diversity patterns in surface bacterioplankton metacommunities. Aquat Microb Ecol 59: 1–10.
Besemer K, Singer G, Quince C, Bertuzzo E, Sloan W, Battin TJ . (2013). Headwaters are critical reservoirs of microbial diversity for fluvial networks. Proc R Soc B 280: 20131760.
Borcard D, Gillet F, Legendre P . (2011) Numerical Ecology with R. Springer Science and Business Media: New York, NY, USA.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621–1624.
Cottrell MT, del Giorgio PA, Kirchman DL . (2015). Biogeography of globally distributed bacteria in temperate and boreal Québec lakes as revealed by tag pyrosequencing of 16S rRNA genes. Aquat Microb Ecol; doi: 10.3354/ame01776.
Crump BC, Adams HE, Hobbie JE, Kling GW . (2007). Biogeography of bacterioplankton in lakes and streams of an Arctic Tundra catchment. Ecology 88: 1365–1378.
Crump BC, Amaral-Zettler LA, Kling GW . (2012). Microbial diversity in arctic freshwaters is structured by inoculation of microbes from soils. ISMEJ 6: 1629–1639.
Crump BC, Hopkinson CS, Sogin ML, Hobbie JE . (2004). Microbial biogeography along an estuarine salinity gradient: combined influences of bacterial growth and residence time. Appl Environ Microb 70: 1494–1505.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Fierer N, Morse JL, Berthrong ST, Bernhardt ES, Jackson RB . (2007). Environmental controls on the landscape-scale biogeography of stream bacterial communities. Ecology 88: 2162–2173.
Fierer N, Jackson RB . (2006). The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103: 626–631.
Fujii M, Kojima H, Iwata T, Urabe J, Fukui M . (2012). Dissolved organic carbon as major environmental factor affecting bacterioplankton communities in mountain lakes of Eastern Japan. Microb Ecol 63: 496–508.
Geobase . (2009). Land Cover, Circa 2000-Vector..
Gibson JJ, Prepas EE, McEachern P . (2002). Quantitative comparison of lake throughflow, residency, and catchment runoff using stable isotopes: modelling and results from a regional survey of Boreal lakes. J Hydrol 262: 128–144.
Gilbert JA, Steele JA, Caporaso JG, Steinbrück L, Reeder J, Temperton B et al. (2012). Defining seasonal marine microbial community dynamics. ISME J 6: 298–308.
Jones S, Cadkin T, Newton R, McMahon K . (2012). Spatial and temporal scales of aquatic bacterial beta diversity. Front Microbiol 3: 318.
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21: 494–504.
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH . (2012). Beyond biogeographic patterns:processes shaping the microbiallandscape. Nat Rev Microbiol 10: 497–506.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A . (2005). Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25: 1965–1978.
Heino J, Tolkkinen M, Pirttilä AM, Aisala H, Mykrä H . (2014). Microbial diversity and community– environment relationships in boreal streams. J Biogeogr 41: 2234–2244.
Langenheder S, Kely AJ . (2011). Species sorting and neutral processes are both important during the initial assembly of bacterial communities. ISME J 5: 1086–1094.
Lapierre JF, del Giorgio PA . (2014). Partial coupling and differential regulation of biologically and photochemically labile dissolved organic carbon across boreal aquatic networks. Biogeosciences 11: 5969–5985.
Lapierre JF, Seekell DA, del Giorgio PA . (2015). Climate and landscape influence on indicators of lake carbon cycling through spatial patterns in dissolved organic carbon. Glob Change Biol 21: 4425–4435.
Lindström ES, Bergström A-K . (2004). Influence of inlet bacteria on bacterioplankton assemblage composition in lakes of different hydraulic retention time. Limnol Oceangr 49: 125–136.
Lindström ES, Forslund M, Algesten G, Bergström A-K . (2006). External control of bacterial community structure in lakes. Limnol Oceangr 51: 339–342.
Lear G, Washington V, Neale M, Case B, Buckley H, Lewis G . (2013). The biogeography of stream bacteria. Global Ecol Biogeog 22: 544–554.
Lindström ES, Langenheder S . (2012). Local and regional factors influencing bacterial community assembly. Environ Microbiol Reports 4: 1–9.
Liu L, Yang J, Yu Z, Wilkinson D . (2015). The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J 9: 2068–2077.
Logares R, Lindström ES, Langenheder S, Logue JB, Paterson H, Laybourn-Parry J et al. (2013). Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J 7: 937–948.
Logue JB, Lindström ES . (2010). Species sorting affects bacterioplankton community composition as determined by 16 S rDNA and 16 S rRNA fingerprints. ISME J 4: 729–738.
Lozupone CA, Knight R . (2007). Global patterns in bacterial diversity. Proc Natl Acad Sci USA 104: 11436–11440.
Magoč T, Salzberg SL . (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27: 2957–2963.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Nelson CE, Sadro S, Melack JM . (2009). Contrasting the influences of stream inputs and landscape position on bacterioplankton community structure and dissolved organic matter composition in high‐elevation lake chains. Limnol Oceangr 54: 1292–1305.
Newton RJ, Jones SE, Eiler A, McMahon KA, Bertilsson S . (2011). A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol R 75: 14–49.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MH, Wagner H . (2015), vegan: Community Ecology Package. R package version 2.3-0 http://CRAN.R-project.org/package=vegan.
Pommier T, Canbäck B, Riemann L, Boström KH, Simu K, Lundberg P et al. (2006). Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol 16: 867–880.
R Core Team . (2013). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria. Available from http://www.R-project.org/.
Read D, Gweon H, Bowes M, Newbold L, Field D, Bailey M, Griffiths R . (2015). Catchment-scale biogeography of riverine bacterioplankton. ISME J 9: 516–526.
Ren L, Jeppesen E, He D, Wang J, Liboriussen L, Xing P et al. (2015). pH influences the importance of niche-related and neutral processes in lacustrine bacterioplankton assembly. Wommack, KE (ed). Appl Environ Microb 81: 3104–3114.
Rasilo T, Prairie YT, del Giorgio PA . (2015). Large-scale patterns in summer diffusive CH4 fluxes across boreal lakes, and contribution to diffusive C emissions. Glob Change Biol 21: 1124–1139.
Ruiz-González C, Niño-García JP, Lapierre JF, del Giorgio PA . (2015a). The quality of organic matter shapes the functional biogeography of bacterioplankton across boreal freshwater ecosystems. Global Ecol Biogeogr 24: 1487–1498.
Ruiz-González C, Niño-García JP, del Giorgio PA . (2015b). Terrestrial origin bacterial communities in complex boreal freshwater networks. Ecol Lett 18: 1198–1206.
Savio D, Sinclair L, Ijaz UZ, Blaschke AP, Reischer GH, Bloeschl G et al. (2015). Bacterial diversity along a 2 600 km river continuum. Environ Microbiol; doi: 10.1111/1462-2920.12886.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al. (2009). Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75: 7537–7541.
Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N . (2013). A meta-analysis of changes in bacterial and archaeal communities with time. ISME J 7: 1493–1506.
Souffreau C, Van der Gucht K, Van Gremberghe I, Kosten S, Lacerot G, Meirelles Lobão L et al. (2015). Environmental rather than spatial factors structure bacterioplankton communities in shallow lakes along a > 6000 km latitudinal gradient in South America. Environ Microbiol 17: 2336–2351.
Székely AJ, Berga ME, Langenheder S . (2012). Mechanisms determining the fate of dispersed bacterial communities in new environments. ISME J 7: 61–71.
Turner KW, Edwards TWD, Wolfe BB . (2014). Characterising runoff generation processes in a Lake-Rich Thermokarst Landscape (Old Crow Flats, Yukon, Canada) using δ 18O, δ 2H and d-excess measurements. Permafrost Periglac 25: 53–59.
Van der Gucht K, Cottenie K, Muylaert K, Vloemans N, Cousin S, Declerck S et al. (2007). The power of species sorting: local factors drive bacterial community composition over a wide range of spatial scales. Proc Natl Acad Sci USA 104: 20404–20409.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 73: 5261–5267.
Wilhelm L, Besemer K, Fragner L, Peter H, Weckwerth W, Battin T . (2015). Altitudinal patterns of diversity and functional traits of metabolically active microorganisms in stream biofilms. ISME J 9: 2454–2464.
Yannarell AC, Triplett EW . (2005). Geographic and environmental sources of variation in lake bacterial community composition. Appl Environ Microb 71: 227–239.
Acknowledgements
We thank Ryan Hutchins for help with GIS analyses, Annick St-Pierre, Alice Parkes and the whole CarBBAS team for their contribution to the field and laboratory components of this research, and Nicolas Fortin St-Gelais for critical comments and discussion. This study is part of the program of the Carbon Biogeochemistry in Boreal Aquatic Systems (CarBBAS) Industrial Research Chair, co-funded by the Natural Science and Engineering Research Council of Canada (NSERC) and Hydro-Québec.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Niño-García, J., Ruiz-González, C. & del Giorgio, P. Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. ISME J 10, 1755–1766 (2016). https://doi.org/10.1038/ismej.2015.226
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.226
This article is cited by
-
High-resolution freshwater dissolved calcium and pH data layers for Canada and the United States
Scientific Data (2024)
-
Climate and Environmental Variables Drive Stream Biofilm Bacterial and Fungal Diversity on Tropical Mountainsides
Microbial Ecology (2024)
-
Effects of Cascade Reservoirs on Spatiotemporal Dynamics of the Sedimentary Bacterial Community: Co-occurrence Patterns, Assembly Mechanisms, and Potential Functions
Microbial Ecology (2024)
-
The different responses of planktonic bacteria and archaea to water temperature maintain the stability of their community diversity in dammed rivers
Ecological Processes (2023)
-
Glacial recession in Andean North-Patagonia (Argentina): microbial communities in benthic biofilms of glacier-fed streams
Hydrobiologia (2023)
