
Abstract
Despite increasing knowledge on phylogenetic composition of the human microbiome, our understanding of the in situ activities of the organisms in the community and their interactions with each other and with the environment remains limited. Characterizing gene expression profiles of the human microbiome is essential for linking the role of different members of the bacterial communities in health and disease. The oral microbiome is one of the most complex microbial communities in the human body and under certain circumstances, not completely understood, the healthy microbial community undergoes a transformation toward a pathogenic state that gives rise to periodontitis, a polymicrobial inflammatory disease. We report here the in situ genome-wide transcriptome of the subgingival microbiome in six periodontally healthy individuals and seven individuals with periodontitis. The overall picture of metabolic activities showed that iron acquisition, lipopolysaccharide synthesis and flagellar synthesis were major activities defining disease. Unexpectedly, the vast majority of virulence factors upregulated in subjects with periodontitis came from organisms that are not considered major periodontal pathogens. One of the organisms whose gene expression profile was characterized was the uncultured candidate division TM7, showing an upregulation of putative virulence factors in the diseased community. These data enhance understanding of the core activities that are characteristic of periodontal disease as well as the role that individual organisms in the subgingival community play in periodontitis.
Similar content being viewed by others

Introduction
It is now recognized that the study of human health must include the role of the microbiome in maintaining homeostasis of the whole human system. The oral microbiome is one of the best characterized human body sites (Socransky et al., 1998; Paster et al., 2001; Marsh, 2006; Haffajee et al., 2008; Belda-Ferre et al., 2012; Peterson et al., 2013), comprising an extremely complex and highly organized biofilm community (Kolenbrander, 2000; Kolenbrander et al., 2002). More than 600 bacterial species have been identified in the oral cavity (Paster et al., 2001; Dewhirst et al., 2010). Many oral bacterial species have not yet been cultivated, and the only information we possess about them derives from their 16S rRNA phylogenetic affiliation.
Periodontitis is an oral polymicrobial disease caused by the coordinated action of a complex microbial community, which results in inflammation and destruction of the periodontium in susceptible hosts. This widespread disease is responsible for half of all tooth loss in adults; it occurs in moderate form in 39% of American adults and in severe form in 9% of adults, with a prevalence of 70% in adults older than 65 years (Eke et al., 2012).
Despite the limited number of studies, deep 16S rRNA sequencing using next-generation sequencing methods has yielded a wealth of knowledge regarding the genetic composition of the oral microbiome in periodontal health and disease (Griffen et al., 2012; Liu et al., 2012; Segata et al., 2012). Using checkerboard DNA–DNA hybridization technique, periodontitis-associated taxa have been cataloged into groups or ‘complexes,’ representing bacterial consortia that appear to occur together and that are associated with various stages of disease (Socransky et al., 1998). The ‘red complex,’ which appears later in biofilm development, comprises three species that are considered the major periodontal pathogens: Porphyromonas gingivalis, Treponema denticola and Tannerella forsythia (Socransky et al., 1998; Holt and Ebersole, 2005).
Despite a good understanding of the association between red complex species and periodontitis, we have only limited information on the in situ activity of these organisms. Cataloguing the activities of each bacterial species in a community may provide more insight into pathogenesis than simple enumeration of that community’s gene content. This is because the community functions as a system, and it is the activities and interactions of the system that control the fate of the microbiome. One approach to describing the functional roles of specific bacteria is to study the community’s metatranscriptome in situ. This technique, although indirect, is more direct than phylogenetic analysis. It has been successfully applied to study various environmental microbial communities (Frias-Lopez et al. 2008; McCarren et al. 2010) and, less frequently, to study human-associated microbiomes (Booijink et al., 2010; Poroyko et al., 2010; Giannoukos et al., 2012; Xiong et al., 2012). To our knowledge, these methods have never been applied to study the microbiome in periodontitis.
Therefore, the goal of the present study was to characterize in situ gene expression patterns of the oral microbiome in periodontal health and disease. We addressed the following questions: What is the function of the expressed genes and particularly those most highly expressed? Do known periodontal pathogens appear to drive a large portion of activity as measured by their gene expression? Among genes that are more highly expressed in periodontitis samples, do virulence factors predominate? Do genes encoding virulence factors tend to be expressed by known periodontal pathogens?
Materials and methods
Study design and subject population
We conducted a cross-sectional comparison of microbial gene expression in subjects with (n=7) and without (n=6) periodontitis who were part of an ongoing study. All aspects of the study protocol were approved by the Institutional Review Board at the Forsyth Institute. The study was described thoroughly to all subjects before obtaining informed consent.
Inclusion criteria were as follows: all study subjects were >18 years of age, had ⩾15 natural teeth and were in good general health. Periodontally healthy subjects had no pockets >3 mm. Periodontitis subjects had ⩾8 teeth with pockets >5 mm and ⩾8 teeth with clinical attachment loss >3 mm.
Exclusion criteria were as follows: subjects were excluded if they were cigarette smokers; were pregnant or nursing; received antibiotic or periodontal therapy in the previous three months; had any systemic condition potentially affecting the course of periodontal disease (for example, diabetes or AIDS); or had any condition requiring antibiotic coverage during periodontal therapy.
Sample collection
After removal of supragingival plaque, subgingival plaque samples were taken separately from each site using individual sterile Gracey curettes and each sample placed in individual tubes containing 1 ml of RNAlater (Life Technologies, Grand Island, NY, USA). For three periodontally healthy subjects, six separate samples from sites with pocket depths ⩽2 mm, no bleeding on probing and no redness were pooled separately (one pool for each subject). For the other three periodontally healthy subjects, only one site was sampled. Samples were initially pooled for fear of insufficient RNA extraction yields. Because we obtained sufficient RNA from single samples, we modified the protocol. For periodontitis subjects, sites with deep pockets (⩾6 mm) that bled on probing were sampled. Four subjects had six sites sampled and pooled separately (one pool per subject), whereas the remaining three subjects contributed with one sample each.
Community DNA and RNA extraction
Cells were collected by centrifugation for 10 min at maximum speed in a microcentrifuge. A quantity of 600 μl of mirVana kit (Life Technologies) lysis/binding buffer and 300 μl of 0.1-mm zirconia-silica beads (BioSpec Products, Bartlesville, OK, USA) were added to the samples. The beads were cleaned and sterilized beforehand with a series of HCl acid and bleach washes. Finally, the beads were treated with diethylpyrocarbonate overnight and autoclaved. Samples were bead beated for 1 min at maximum speed. DNA and RNA were extracted simultaneously following the protocol of mirVana Isolation kit for RNA and ToTALLY RNA kit (Life Technologies) for DNA. Eukaryotic DNA was removed using the MolYsis kit (Molzym GmbH & Co. KG, Bremen, Germany). MICROBioEnrich (Life Technologies) was used to remove eukaryotic RNA and MICROBExpress to remove prokaryotic rRNA. All kits were used following manufacturer’s instructions.
DNA, RNA amplification and Illumina sequencing
DNA amplification was performed using the Illustra GenomiPhi V2 amplification kit (GE Healthcare Life Sciences, Piscataway, NJ, USA) according to manufacturer’s instructions. RNA amplification was performed on total bacterial RNA using MessageAmp II-Bacteria RNA amplification kit (Life Technologies) following the manufacturer’s instructions. Sequencing was performed at the Forsyth Institute and at the Biopolymers Facility Harvard Medical School. At the Forsyth Institute, for RNA sequencing, Illumina adapter-specific primers (Illumina, San Diego, CA, USA) were used to amplify and selectively enrich for the cDNA generated from enriched mRNA. Quantified libraries were pooled and sequenced using the Miseq v2, 2 × 150 cycle cartridge (Illumina). The Nextera XT kit was used to generate libraries from amplified DNA. Normalized libraries were pooled and sequenced using the 2 × 250 Miseq v2 cartridge. At the Biopolymers Facility Harvard Medical School, samples were prepped using NuGen’s mRNA-seq kit (Illumina) into next-gen libraries with Illumina adapters. The libraries were then clustered on a single-read flow-cell using Illumina’s cBot and the sequencing run for 100 cycles single-end for the DNA samples and 50 cycles paired-end for RNA samples on the Illumina HiSeq 2500 (Illumina).
Selection of genomes in databases
Genomes of archaea and bacteria and their associated information were downloaded from the Human Oral Microbiome Database database server (http://www.homd.org/), the Pathosystems Resource Integration Center (PATRIC) ftp server (http://www.patricbrc.org/portal/portal/patric/Home) (Wattam et al., 2013) and the J Craig Venter Institute (http://www.jcvi.org). Viral genomes were downloaded from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/Viruses/all.fna.tar.gz). A total of 498 genomes from 283 species of bacteria and 2 genomes from 1 archaea species were used in the analysis (Supplementary Table 1). A total of 4000 viral genomes was used to create the viral database.
Comparison of different alignment algorithms on in silico communities
We used the simulator ‘wgsim’ (https://github.com/lh3/wgsim), which creates sequences that mimic results obtained by Illumina sequencing, to generate sequences from the oral genomic database consisting of 500 oral microbial genomes (Supplementary Table 1). We generated 200 000 reads with sizes equal to those from the actual results (see Supplementary Table 2 for running parameters). We mapped those reads against the database used in our experiments, comparing the following seven algorithms to identify the one that most accurately recruited the most reads: bowtie2, stampy, bowtie, gassst, bwa, smalt and ssaha2, on the ‘in silico’ community. After mapping with the different algorithms, we evaluated the results with the script 'wgsim_eval.pl' from the same program.
Short reads sequence alignment analysis
Low-quality sequences were removed from the query files. ‘fast_clipper' and ‘fastq_quality_filter' from the Fastx-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) were used to remove short sequences with quality score >20 in >80% of the sequence. For paired-end sequences, we then selected pairs present in both files. We also removed prokaryotic ribosomal RNA sequences and human sequences (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/chromosomes) bioinformatically. Cleaned files were then aligned against the bacterial/archaeal database using bowtie2. We generated a.gff file to map hits to different regions in the genomes of our database. Read counts from the SAM files were obtained using bedtools multicov from bedtools (Quinlan and Hall, 2010).
Phylogenetic analysis of the metagenome and metatranscriptome
Counts from the DNA and RNA libraries were used to determine the phylogenetic composition of the respective libraries. We created a.gff file containing information on whole genomes that was used to assign hits to genomes. Counts were normalized by dividing by the total number of reads for each sample and log-transforming the resulting fractions (Ramette, 2007; Brown et al., 2011). Normalized phylogenetic profiles were used for principal coordinates analysis with Manhattan distance performed using the R BiodiversityR package (Kindt and Coe, 2005). In addition, we performed the non-parametric test included in the NOISeq package (Tarazona et al., 2011) on the raw counts assigned to the different species to determine significant changes in counts at the species level.
Differentially expressed genes in subjects with vs without periodontitis
For assessing differential expression in genes within a specific species, we normalized the transcript counts by the relative frequency of the species in the metagenome database. In the case of Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, we did not normalized enrichment analysis by relative abundance, as we were treating the whole community as a single organism.
To identify differentially expressed genes from the RNA libraries, we applied non-parametric tests to the normalized counts using NOISeq’s default conditions (k=0.5, pnr=0.2, nss=5, v=0.02, lc=1, replicates=‘biological’) and Trimmed Mean of M-values (‘TMM' option) using the threshold value for significance suggested by the authors (Tarazona et al., 2011; Soneson and Delorenzi, 2013).
We obtained a heat-map representation of the expression profiles for the 506 most highly expressed genes across samples as well as the profile corresponding to 1000 significantly differentially expressed genes according to the NOISeq results that showed the highest differences in expression. Counts for those genes were first normalized according to size and the BiodiversityR package (Kindt and Coe, 2005) was used to χ2 transform the frequencies (Legendre and Gallagher, 2001). Using the ‘heatmap.2' function in R, we clustered samples and represented their heat-maps based on the expression profiles. The clustering function for ‘heatmap.2' was ‘hclust' selecting ‘complete' (complete-linkage) as clustering method.
GO and KEGG pathway enrichment analysis
To evaluate functional activities differentially represented in health or disease, we mapped the differentially expressed genes to known biological ontologies based on the GO project (http://www.geneontology.org/) and the KEGG metabolic pathways database (http://www.genome.jp/kegg/). GO terms and KEGG pathways to which the different open reading frames (ORFs) belong were obtained from the PATRIC database (http://patricbrc.org/portal/portal/patric/Home). GO terms and KEGG pathways from genomes not present in the PATRIC database and whose annotation was obtained from the Human Oral Microbiome Database database or J Craig Venter Institute were acquired using the program blast2GO under the default settings (Götz et al., 2008).
Enrichment analysis on these sets was performed using the R package ‘GOseq,' which accounts for biases because of over-detection of long and highly expressed transcripts (Young et al., 2010). Gene sets with ⩽10 genes were excluded from the analysis. We used the REVIGO web page (Supek et al., 2011) to summarize and remove redundant GO terms from the results. Only GO terms with false discovery rate <0.05 were used. REVIGO plots were obtained for two GO categories: biological processes and molecular functions. The GO project describes a biological process as a recognized series of events or molecular functions with a defined beginning and end, and molecular functions as activities that occur at the molecular level, such as catalytic or binding activities.
Quantification of putative virulence factors
To identify putative virulence factors, we used the Virulence Factors of Pathogenic Bacteria Database (VFDB; http://www.mgc.ac.cn/VFs/). Others have used a similar approach to identify putative virulence factors in genomic islands (Ho Sui et al., 2009). The VFDB contains 1143 virulence factors and 5927 virulence factor-related genes from 75 pathogenic bacterial genera (Chen et al., 2012). We performed a BLAST similarity search of encoded proteins from the genomes in our database against the VFDB, with an e-value cutoff of 10−25 and identity >99% to exclude distant homologs.
Results
In the comparison of algorithms for recruiting metatranscriptomic reads, ‘bowtie2' performed the best using either single-end reads (98.2 reads mapped correctly) or paired-end reads (96.9 reads mapped correctly; Supplementary Table 3).
Phylogenetic comparison of the metagenome and metratranscriptome in health and disease
Sequencing yields ranged from 0.4 to 20 million and 0.9 to 39.0 million for the metagenomes and metatranscriptomes, respectively (Supplementary Table 4). No viral sequences were identified in either set of libraries. Of the 1 278 494 genes contained in the genomic database, 702 106 had ⩾1 hit in ⩾1 sample.
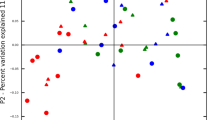
Principal coordinates analysis of the phylogenetic profiles based on either the metagenome or metatranscriptome did not clearly distinguish the subjects with and without periodontitis. Nonetheless, six of the seven metatranscriptome of samples with periodontitis clustered along principal coordinates analysis-1, which captured most of the variance (Figure 1).

Principal coordinate analysis (PCoA) of phylogenetic profiles from metagenomes and metatranscriptomes. Ordination graph for the first two axes of a PCoA of the phylogenetic profiles obtained from the metagenome and metatranscriptome of the oral microbial communities in healthy sites and sites with severe periodontitis. The % on the axes represents the variance explained by each of the coordinates.
After ranking the species based on fold changes in hits with vs without periodontitis, P. gingivalis, T. forsythia and T. denticola exhibited higher mean abundance and higher levels of gene expression in periodontitis than in healthy subjects (Figure 2 and Supplementary Table 5). However, the increase in numbers of other species was not always accompanied by an increase in inferred activity and vice versa. For instance Acinetobacter baumannii was significantly more abundant in periodontitis than in healthy subjects but did not have high levels of gene expression in disease, whereas others, such as Bacteroidetes oral taxon 274, Corynebacterium matruchotii or Leptotrichia hofstadii, showed a decrease in relative numbers but represented a high proportion of the metatranscriptome library (Figure 2).

Rank distribution of statistically significant relative increase in number of hits for the metagenome and metatranscriptome results. The ratio of counts in disease vs health was log2 transformed and plotted according to ranks. The statistical significance was calculated using the non-parametric test implemented in the program NOISeq as described in the Materials and methods section. Only species with significant differences, according to NOISeq, either in metagenomic or metatranscriptomic counts, are presented. In green, species with statistical differences in both metagenome and metatranscriptome. In blue, species with statistical differences in metagenomic counts. In red, species with statistical differences in metatranscriptome counts. Red star indicates major periodontal pathogens previously assigned to the ‘red-complex.’
We compared phylogenetic assignment of expression levels of differentially expressed genes in the non-pooled vs pooled samples after trimmed mean of M-values normalization and we observed similar numbers for the species with higher differences in expression (Supplementary Figure 1). All members of the ‘red- complex’ were represented in both set of samples showing similar numbers of differentially expressed genes. Furthermore, other organisms that we found important both in numbers as well as in expression of virulence factors (for example, Corynebacterium matruchotii, see Supplementary Figure 1) showed also similar patterns in both set of samples.
Global patterns of gene expression in health and disease: enrichment of GO terms and KEGG metabolic pathways
In total, NOISeq analysis identified 91 113 differentially represented genes without normalizing for species abundance and 123 317 differentially expressed genes after normalization by relative abundance of the different species in the community. We analyzed the expression profiles of the most highly represented genes across samples (Figure 3a) as well as the profiles for the most highly differentially represented genes in our libraries (Figure 3b). Although both profiles separated clusters from healthy and periodontitis samples, the profiles for the most highly differentially expressed genes better distinguished the two subject groups. Expression profiles of the most highly differentially represented genes seem to be highly similar across samples in the periodontitis group. If confirmed, this observation would suggest that periodontitis might be characterized by a core of specific microbial activities (Figure 3b).

Heat maps of gene expression profiles in the metatranscriptomes. (a) Profiles of the 1212 most highly expressed genes across samples. Genes were first normalized by gene length and χ2 transformed before analysis. Most highly expressed genes were selected according to the sum of normalized counts across samples. (b) Profiles of the 1000 most highly differentially expressed genes across samples. Genes were selected from the NOISeq results and counts were χ2 transformed before analysis. Color bars in red correspond to healthy samples and in green to periodontitis samples.
A total of 146 GO terms were overrepresented and 42 underrepresented in healthy vs periodontitis subjects (Supplementary Table 6). Using the same package, ‘GOseq,' but mapping to KEGG pathways instead of GO categories, we identified 14 pathways overrepresented and 18 underrepresented in disease (Supplementary Table 6). There were no meaningful differences in results for pooled and un-pooled samples. In particular, the themes that emerged from GO analysis were the same regardless of pooling of samples (Supplementary Figure 2). The differences we observed when splitting the samples into the two groups may be due in part to the reduction in the number of samples per group. In the whole analysis, we compared six healthy vs seven periodontitis samples, whereas in Supplementary Figure 2 we compared three pooled and four non-pooled samples separately, reducing the statistical power of the analysis.
GO biological processes related to flagellar motility, peptides transport, iron acquisition and beta-lactam degradation were overrepresented in disease, as was biosynthesis of the lipid A component of endotoxins from Gram-negative bacteria (Figures 4a and b). GO biological processes underrepresented in disease included potassium transport and polysaccharide biosynthesis. From gene set enrichment analysis based on KEGG pathways, which represents a completely different ontology, the overrepresented pathways were related to broad activities, such as energy metabolism (for example, Aminoacyl-tRNA biosynthesis, nitrogen metabolism), carbohydrate metabolism (for example, glycolysis/gluconeogenesis, citrate cycle, pyruvate metabolism), bacterial motility and chemotaxis, and genes that function in electron transport (for example, photosynthesis and porphyrin and chlorophyll metabolism; Supplementary Table 6). Biosynthesis of secondary metabolites and antibiotics, metabolism of terpenoids and alkaloids and lipid metabolism accounted for most of the KEGG pathways underrepresented in the periodontitis samples (Supplementary Table 6).

GO enrichment analysis summarized and visualized as a scatter plot using REVIGO. (a) Summarized GO terms related to biological processes in periodontitis. (b) Summarized GO terms related to biological processes in health. (c) Summarized GO terms related to molecular function in periodontitis. (d) Summarized GO terms related to molecular function in health. GO terms are represented by circles and are plotted according to semantic similarities to other GO terms (adjoining circles are most closely related). Circle size is proportional to the frequency of the GO term, whereas color indicates the log10 P value (red higher, blue lower).
Expression profiles of putative virulence factors in major periodontal pathogens
The three members of the red complex showed high expression of metalloproteases and peptidases (Supplementary Table 7, in red). In addition, proteins involved in iron metabolism represented an important fraction of upregulated putative virulence factors in these periodontal pathogens. These included ferrous iron transport protein B, iron compound ABC transporter ATP-binding proteins, ferric enterobactin transport ATP-binding protein FepC and iron chelate uptake ABC transporter FeCT permease proteins (Supplementary Table 7, in green). In periodontitis subjects, there was higher expression of P. gingivalis hemolysin genes and P. gingivalis and T. denticola genes involved in vitamin B12 import (for example, vitamin B12 ABC transporter permease component BtuC).
Also more highly expressed in periodontitis samples were genes encoding other proteins not involved directly in iron metabolism, such as ATP-dependent Clp protein, ATP-binding subunit ClpA, ClpB protein; the chaperone ClpB protein, described as involved in cellular invasion in P. gingivalis; GroEL; protein export cytoplasm secA ATPase RNA helicase and RNA polymerase sigma factor RpoD.
Most of the genes needed to synthesize flagella in T. denticola were overrepresented in the periodontitis metratranscriptome. These included flagellar basal-body FlgC, FlgF and FlgG, flagellar hook proteins FlgE and FlgK, flagellar motor rotation proteins MotA and MotB, flagellar motor switch proteins FliG and FliM, flagellin protein FlaA, flagellar M-ring protein FliF, flagellum-specific ATP synthase FliI and proteins involved in flagella biosynthesis (FleN, FlhA-B, FlhF and FliP; Supplementary Table 7). Of the three major periodontal pathogens, T. denticola was the only one exhibiting over-representation of proteins involved in oligopeptide transport (OppA, OppD and OppF).
Expression of virulence factors and virulence factor-related genes in the oral community
A total of 7846 hits of putative virulence factors from 255 species were identified in the genes overrepresented in periodontitis samples, although for only 64 species did this include ⩾40 virulence factors. Members of the ‘red-complex’ as well as Aggregatibacter actinomycetemcomitans, an organism associated with localized juvenile periodontitis, over-expressed a large number of the putative virulence factors in the periodontal samples (Figure 5). Nonetheless, surprisingly enough, a set of organisms that have been previously associated only with diseases other than periodontitis or with periodontally healthy sites, such as Neisseria spp., Corynebacterium matruchotii, Rothia dentocariosa, Veillonella parvula, Actinomyces spp. and others, over-expressed a large number of putative virulence factors that could have importance in the evolution of the disease (Figure 5). Moreover, a large number of different homologs from different ORFs within each genome were all over-expressed. For instance, C. matruchotii showed upregulation of seven different ORFs with homology to the Fe(3+) ions import ATP-binding protein FbpC and 10 ORFs homologs of iron (III) ABC transporter ATP-binding proteins in the VFDB. Moreover, C. matruchotii upregulated 20 different ORFs that are annotated as putative hemagglutinin hemolysin-related proteins (Supplementary Table 8, in green).

Ranked species by the number of upregulated putative virulence factors in the metatranscriptome. Putative virulence factors were identified by alignment of the protein sequences from the different genomes against the Virulence Factors of Pathogenic Bacteria Database as described in the Materials and methods section. Number in the graph are the absolute number of hits for the different species for the putative virulence factors identified. In red are the members of the ‘red-complex.’
There were several other pathways for which genes were overexpressed in periodontitis samples in a large number of species. Second in number only to those related to iron metabolism were genes in lipopolysaccharide biosynthesis pathways (Supplementary Table 8, in red). Other such pathways or groups included: oxidative stress tolerance (trigger factor, superoxide dismutase and alkyl hydroperoxide reductase C protein AhpC); specific regulatory proteins (for example, phosphate regulon transcriptional regulatory protein PhoB SphR, RNA polymerase sigma factors RpoD/RpoD and translation elongation factor Tu); specialized protein transport systems (for example, zinc ABC transporters ZnuA and ZnuC and ABC-type multi-drug transport systems ATPase and permease components); oligopeptide ABC transporters; nitrate reductase proteins (alpha, beta, delta and gamma subunits); serine-proteases (for example, MucD/AlgY, DegP/HtrA); hemolysins; large numbers of proteins involved in flagellar biosynthesis (for example, FlaA-B, FlaG, FleN, FlgB-I, FlgK-L, FlhA-B, FlhF, FliD-I, FliK-N, FliP-S, MotA and MotB) and chaperones such as GroEL. Organisms expressing high numbers of putative virulence factors expressed high levels of a putative ABC transporter that acts as an efflux pump of the anticancer drug daunorubicin (DrrA), as well as the corresponding daunorubicin-resistant ABC transporter ATPase subunit.
The role of non-cultivable organisms in health and disease: the case of candidate division TM7
Despite the sequence of candidate division TM7 genome being incomplete, we identified differentially expressed genes, including cell division proteins (for example, chromosomal replication initiator proteins DnaA, FtsA and FtsH) and chaperones (GroEL, GroES, DnaJ and Hsp20). In addition, several putative virulence factors were also more highly expressed in periodontitis samples. These were related to iron acquisition (for example, hemagglutinin-related proteins and an exoprotein involved in heme utilization of adhesion), pili biosynthesis (PilB and PilC) and oligopeptide transport (OppA; Supplementary Table 9). OppA was also highly overrepresented in the periodontopathogen T. denticola.
Discussion
To gain insight into the functional phenotype of periodontal dysbiosis, we used a combined metagenomic/metatranscriptomic approach. This allowed us to infer functional differences between the subgingival microbiota of periodontal health and disease and generate hypotheses regarding the role of the different community members in controlling the fate of the biofilm. We took advantage of both next generation sequencing techniques and the fact that a large number of genomes from oral isolates have been sequenced, although not all (Chen et al., 2010). Moreover, the amplification methods used for metagenomic and metatranscriptomic analysis do not introduce bias in the final relative proportions of hits (Teles et al., 2007; Hoeijmakers et al., 2011).
In a recent deep sequencing analysis using 16S rDNA, members of the genus Prevotella, Fusobacterium, Treponema, Selenomonas and Porphyromonas were more abundant, whereas Actinomyces and Streptococcus were less abundant in samples from periodontitis compared with healthy subjects (Griffen et al., 2012). We observed similar patterns using metagenomic analysis, although with a slight increase in numbers of Streptococcus in periodontitis samples. Moreover, we observed high numbers of members of the genus Neisseria in the diseased samples and of candidate division TM7 in samples from both healthy and periodontitis sites. Two recent studies have reported a higher number of these organisms in oral samples (Liu et al., 2012; Segata et al., 2012) contradicting previous studies that had shown high prevalence but with very low abundances (Brinig et al., 2003; Podar et al., 2007), probably due to an improvement in detection techniques. We also looked at the presence of viruses and archaea, which have been previously associated with disease (Lepp et al., 2004; Slots, 2010). Archaea accounted for a very small fraction of the total community, which agrees with other reports on the abundances of these organisms in oral samples (Lepp et al., 2004) and we did not detect any viral sequence in either the metagenome or the metatranscriptome.
Expression profiles of the most highly differentially expressed genes seem to be highly similar across samples in the periodontitis group. If confirmed, this observation would suggest that periodontitis might be characterized by a core of specific microbial activities.
Testing of over-representation of GO terms and KEGG metabolic pathways in the microbial community as a meta-organism gives an overall view of the microbial activities under different states of health. The results pointed to several functional signatures characteristic of periodontitis. Iron acquisition was abundantly overrepresented in the periodontitis samples. Iron is an essential enzymatic cofactor and microbial pathogens have developed a large number of strategies to acquire it (Brochu et al., 2001; Rhodes et al., 2007; Nairz et al., 2010). Also oligopeptide transport proteins were over-expressed. Some important oral bacteria, such as Parvimonas micra, Fusobacterium nucleatum and P. gingivalis, use peptides more efficiently than free amino acids (Tang-Larsen et al., 1995) and it is plausible that the degree of efficiency in using peptides may influence the ecology of the periodontal biofilm. Lipid-A biosynthesis was also overrepresented in disease. Lipopolysaccharide is a key factor in the development of periodontitis (Fives-Taylor et al., 1999; Jain and Darveau, 2010) and high levels of lipopolysaccharide and lipid A from P. gingivalis have been reported to delay neutrophil apoptosis and provide a mechanism to modulate the restoration and maintenance of inflammation in periodontal tissues (Hiroi et al., 1998; Preshaw et al., 1999; Murray and Wilton, 2003).
The above-described metatranscriptomic differences between subjects with and without periodontitis are consistent with the current understanding of periodontal pathogenesis. A novel observation was the relatively abundant expression in periodontitis samples of genes related to the response to antibiotics in general, and beta-lactam antibiotic degradation processes in particular. Beta-lactamase-producing bacteria have been frequently isolated from the oral cavity (Handal et al., 2004; Rams et al., 2012) and beta-lactamase activity has been observed in adult periodontitis at low-level enzymatic activity but with high prevalence (Van Winkelhoff et al., 1997). Beta-lactamase activity seems to be a frequent phenomenon in samples from polymicrobial diseases (Brook, 2009) and has been detected in specimens of clinical abscesses and mixed infections, such as empyema (Bryant et al., 1980), cerebrospinal fluid (Boughton, 1982) and abscesses (Brook, 1986). What role this enzymatic activity has on the homeostasis of the community is an open question given that none of the samples came from patients being treated with antibiotics at the time of sampling. Nonetheless, the presence and prevalence of beta-lactamase genes in the subgingival microbial community, even in the absence of antibiotic, has public health implications given the rise of resistance among a large number of bacterial pathogens. Our findings suggest that the target of beta-lactamase activity may be a different molecule, which may hamper efforts to eliminate beta-lactamase genes from the genetic pool of the human microbiome.
We also found an under-representation of antibiotic biosynthesis genes in periodontitis samples, consistent with the expression of antibiotics as a potential mechanism of homeostasis in health-associated microbiota. Further, the production of beta-lactamase might represent a mechanism that confers protection to some species in the subgingival microbiota against antibiotics released by others.
P. gingivalis, T. denticola and T. forsythia are considered major periodontal pathogens and appear in great numbers later in biofilm development when the polymicrobial infection is well established (Socransky et al., 1998; Ximenez-Fyvie et al., 2000; Holt and Ebersole, 2005). Consistent with their postulated role, in subjects with severe periodontitis, we observed relatively high levels of expression of proteolysis-related genes. Gingipains or fimbrial genes of P. gingivalis have been described as essential in the early stages of colonization (Imamura, 2003; Enersen et al., 2013); however, we observed no over-representation of these genes in our results, may be due to the fact that we are analyzing severe periodontitis samples and no early stages of the disease.
All members of the ‘red-complex’ exhibited an over-representation of RNA-encoding proteins involved in invasion of cells, including ClpB, a general protection factor against stress, GroEL and DnaK, which have also been associated with cellular invasion (Rodrigues and Progulske-Fox, 2005; Xia et al., 2007; Yuan et al., 2007), and ferritin, linked to invasion of cells by P. gingivalis (Rodrigues and Progulske-Fox, 2005; Xia et al., 2007). GroEL has been identified as a virulence factor in the periodontal pathogen Actinobacillus actinomycetemcomitans (Goulhen et al. 1998).
T. forsythia has been detected intra-cellularly in buccal and crevicular epithelium of patients with periodontitis (Rudney et al., 2005) and has been shown to invade epithelial cells in vitro (Inagaki et al., 2006; Mishima and Sharma, 2011). We observed over-representation in periodontitis of a homolog of the cell-surface-associated protein sialidase NanH (locus VBITanFor42681_2003) required for attachment to epithelial cells (Honma et al., 2011).
T. forsythia and T. denticola over-expressed homologs to internalins, surface proteins that are key in invasion of mammalian cells by Listeria monocytogenes (Lecuit et al., 1997; Cossart et al., 2003), and could have a similar role in those periodontal pathogens.
Finally, we identified as upregulated most of the genes necessary to synthesize flagella in T. denticola as well as genes necessary for chemotaxis that could direct flagellar movement. Flagella are essential virulence factors in T. denticola, the lack of which prevents it from penetrating periodontal tissue (Lux et al., 2001). Taken together, these observations strongly support the idea that in deep bleeding pockets the members of the ‘red-complex’ are actively invading host cells.
Recently, gaining traction is the idea of the ‘pathogenic microbial community’ (Berezow and Darveau, 2011) or ‘the community as pathogen’ (Relman, 2012), which postulates that the integrated actions of the components of the microbial community would result in disease. In part to address this hypothesis, we examined the possible role of organisms not previously recognized as periodontal pathogens. A large number of organisms in the oral community were expressing putative virulence factors. Members of the genus Streptococcus such as S. mitis, generally associated with health, were expressing homologs of virulence factors from known pathogenic streptococci such as; neuraminidase A (Jedrzejas, 2001), zinc metalloprotease ZmpB (Bender and Weiser, 2006) and the saliva-binding protein adhesin B (Herbert et al., 2004).
Surprisingly, C. matruchotii, R. dentocariosa, Veillonella parvula and Neisseria sicca were expressing a large variety of putative virulence factors. Although C. matruchotii and R. dentocariosa have occasionally been associated with disease (Siqueira et al., 2000; Thomas et al., 2012), in general, they (and other Gram-positive organisms) have been considered associated with health of the periodontium (Kolenbrander, 2000; Gross et al., 2010; Liu et al., 2012).
Some of the observed associations point to possible synergistic interactions among community members. Specifically, siderophores such as entorobactins and ABC-type cobalamin Fe3+-siderophores were highly over-represented in the periodontitis samples, yet members of the ‘red-complex’ do not appear to produce them (Brochu et al., 2001; Rhodes et al., 2007). The production of these molecules by the accompanying community may facilitate iron acquisition by the ‘red-complex.’
Finally, genes involved in stress tolerance were highly over-represented in a large fraction of the community and their upregulation may be related to the production of reactive oxygen species during disease (Wright et al., 2011; Bullon et al., 2012).
One important unanswered question in microbiology is the role of uncultured organisms in the activities of the oral community. Candidate bacterial phylum TM7 has no cultivated members and has been detected in high number in oral samples (Brinig et al., 2003; Kumar et al., 2003; Ouverney et al., 2003; Colombo et al., 2009; Liu et al., 2012; Casarin et al., 2013; Kim and Lee, 2013). We still do not know if TM7 has a role in the progression of periodontitis but we identified several homologs to virulence factors expressed in TM7 that were associated with periodontitis. These included the oligopeptide ABC transporter periplasmic oligopeptide-binding protein (OppA), which has been identified as a virulence factor in group A Streptococci (Wang et al., 2005); a hemagglutinin-related protein (locus VBICanDiv80122_0315); exoproteins involved in heme utilization or adhesion (locus VBICanDiv80122_0453); and an homolog of LemA (locus VBICanDiv80122_0048), a widely conserved two-component regulatory system that has been identified as regulating virulence factors and toxin production in Pseudomonas syringae (Kitten et al., 1998).
Also differentially expressed more highly in periodontitis samples were TM7 genes encoding a chitinase (locus VBICanDiv80122_0016) and a putative ROK-family transcriptional regulator (locus VBICanDiv80122_0244), which regulates chitinase expression in the Gram-positive pathogen Listeria monocytogenes at the post-transcriptional level (Larsen et al., 2010). Representatives of both Gram-positive and Gram-negative bacterial pathogens encode chitinases that support infection of non-chitinous mammalian hosts (Frederiksen et al., 2013), possibly by suppressing host innate immunity (Chaudhuri et al., 2013). Finally, we also observed over-representation of genes involved in pili biosynthesis. Although type IV pili may facilitate the adherence of bacteria to epithelial cells, it has been suggested that in the case of TM7 it may be involved in gliding motility (Marcy et al., 2007). These comparative expression studies provide only circumstantial evidence of a pathogenic role of TM7 members; however, they also demonstrate the usefulness of in situ metatranscriptomic analysis for investigating the possible roles of uncultured organisms and for developing new hypothesis to be tested in the laboratory.
Conclusions
Our findings highlight the usefulness of metatranscriptomic analysis for elucidating the potential importance of virulence factor-expressing organisms not previously linked to pathogenesis, also including previously uncultured organisms. Moreover, these results support the idea that the whole community, and not just a selected group of pathogens, promote virulence factors during disease development. The data presented here illustrate the value of using metatranscriptomic analysis to examine microbial activities in whole human microbial communities and as a hypothesis-generating strategy based on results obtained under in situ conditions.
Data accessibility
The data sets used in these analyses were deposited at the Human Oral Microbiome Database under the submission number 20130522 (ftp://ftp.homd.org/publication_data/20130522/).
References
Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, Romero H, Simón-Soro A, Pignatelli M et al. (2012). The oral metagenome in health and disease. ISME J 6: 46–56.
Bender MH, Weiser JN . (2006). The atypical amino-terminal LPNTG-containing domain of the pneumococcal human IgA1-specific protease is required for proper enzyme localization and function. Mol Microbiol 61: 526–543.
Berezow AB, Darveau RP . (2011). Microbial shift and periodontitis. Periodontol 2000 55: 36–47.
Booijink CCGM, Boekhorst J, Zoetendal EG, Smidt H, Kleerebezem M, de Vos WM . (2010). Metatranscriptome analysis of the human fecal microbiota reveals subject-specific expression profiles, with genes encoding proteins involved in carbohydrate metabolism being dominantly expressed. Appl Environ Microbiol 76: 5533–5540.
Boughton WH . (1982). Rapid detection in spinal fluid of beta-lactamase produced by ampicillin-resistant Haemophilus influenzae. J Clin Microbiol 15: 1167–1168.
Brinig MM, Lepp PW, Ouverney CC, Armitage GC, Relman DA . (2003). Prevalence of bacteria of division TM7 in human subgingival plaque and their association with disease. Appl Environ Microbiol 69: 1687–1694.
Brochu V, Grenier D, Nakayama K, Mayrand D . (2001). Acquisition of iron from human transferrin by Porphyromonas gingivalis: a role for Arg- and Lys-gingipain activities. Oral Microbiol Immunol 16: 79–87.
Brook I . (1986). Presence of beta-lactamase-producing bacteria and beta-lactamase activity in abscesses. Am J Clin Pathol 86: 97–101.
Brook I . (2009). The role of beta-lactamase-producing-bacteria in mixed infections. BMC Infect Dis 9: 202.
Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N et al. (2011). Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One 6: e25792.
Bryant RE, Rashad AL, Mazza JA, Hammond D . (1980). Beta-lactamase activity in human pus. J Infect Dis 142: 594–601.
Bullon P, Cordero MD, Quiles JL, Ramirez-Tortosa MDC, Gonzalez-Alonso A, Alfonsi S et al. (2012). Autophagy in periodontitis patients and gingival fibroblasts: unraveling the link between chronic diseases and inflammation. BMC Med 10: 122.
Casarin RCV, Barbagallo A, Meulman T, Santos VR, Sallum EA, Nociti FH et al. (2013). Subgingival biodiversity in subjects with uncontrolled type-2 diabetes and chronic periodontitis. J Periodont Res 48: 30–36.
Chaudhuri S, Gantner BN, Ye RD, Cianciotto NP, Freitag NE . (2013). The Listeria monocytogenes ChiA chitinase enhances virulence through suppression of host innate immunity. MBio 4: e00617-12.
Chen L, Xiong Z, Sun L, Yang J, Jin Q . (2012). VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res 40: D641–D645.
Chen T, Yu W-H, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE . (2010). The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010: baq013.
Colombo APV, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD et al. (2009). Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol 80: 1421–1432.
Cossart P, Pizarro-Cerdá J, Lecuit M . (2003). Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol 13: 23–31.
Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu W-H et al. (2010). The human oral microbiome. J Bacteriol 192: 5002–5017.
Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ . (2012). Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res 91: 914–920.
Enersen M, Nakano K, Amano A . (2013). Porphyromonas gingivalis fimbriae. J Oral Microbiol 5: 20265–20275.
Fives-Taylor PM, Meyer DH, Mintz KP, Brissette C . (1999). Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol 2000 20: 136–167.
Frederiksen RF, Paspaliari DK, Larsen T, Storgaard BG, Larsen MH, Ingmer H et al. (2013). Bacterial chitinases and chitin-binding proteins as virulence factors. Microbiology (Reading, Engl) 159: 833–847.
Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW et al. (2008). Microbial community gene expression in ocean surface waters. Proc Natl Acad Sci USA 105: 3805–3810.
Giannoukos G, Ciulla DM, Huang K, Haas BJ, Izard J, Levin JZ et al. (2012). Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol 13: R23.
Götz S, García-Gómez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36: 3420–3435.
Goulhen F, Hafezi A, Uitto VJ, Hinode D, Nakamura R, Grenier D et al. (1998). Subcellular localization and cytotoxic activity of the GroEL-like protein isolated from Actinobacillus actinomycetemcomitans. Infect. Immun 66: 5307–5313.
Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK et al. (2012). Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J 6: 1176–1185.
Gross EL, Leys EJ, Gasparovich SR, Firestone ND, Schwartzbaum JA, Janies DA et al. (2010). Bacterial 16S sequence analysis of severe caries in young permanent teeth. J Clin Microbiol 48: 4121–4128.
Haffajee AD, Socransky SS, Patel MR, Song X . (2008). Microbial complexes in supragingival plaque. Oral Microbiol Immunol 23: 196–205.
Handal T, Olsen I, Walker CB, Caugant DA . (2004). Beta-lactamase production and antimicrobial susceptibility of subgingival bacteria from refractory periodontitis. Oral Microbiol Immunol 19: 303–308.
Herbert MA, Beveridge CJE, Saunders NJ . (2004). Bacterial virulence factors in neonatal sepsis: group B streptococcus. Curr Opin Infect Dis 17: 225–229.
Hiroi M, Shimojima T, Kashimata M, Miyata T, Takano H, Takahama M et al. (1998). Inhibition by Porphyromonas gingivalis LPS of apoptosis induction in human peripheral blood polymorphonuclear leukocytes. Anticancer Res 18: 3475–3479.
Ho Sui SJ, Fedynak A, Hsiao WWL, Langille MGI, Brinkman FSL . (2009). The association of virulence factors with genomic islands. PLoS One 4: e8094.
Hoeijmakers WAM, Bártfai R, Françoijs K-J, Stunnenberg HG . (2011). Linear amplification for deep sequencing. Nat Protoc 6: 1026–1036.
Holt SC, Ebersole JL . (2005). Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the ‘red complex’, a prototype polybacterial pathogenic consortium in periodontitis. Periodontology 2000 38: 72–122.
Honma K, Mishima E, Sharma A . (2011). Role of Tannerella forsythia NanH sialidase in epithelial cell attachment. Infect Immun 79: 393–401.
Imamura T . (2003). The role of gingipains in the pathogenesis of periodontal disease. J Periodontol 74: 111–118.
Inagaki S, Onishi S, Kuramitsu HK, Sharma A . (2006). Porphyromonas gingivalis vesicles enhance attachment, and the leucine-rich repeat BspA protein is required for invasion of epithelial cells by ‘Tannerella forsythia’. Infect. Immun 74: 5023–5028.
Jain S, Darveau RP . (2010). Contribution of Porphyromonas gingivalis lipopolysaccharide to periodontitis. Periodontol 2000 54: 53–70.
Jedrzejas MJ . (2001). Pneumococcal virulence factors: structure and function. Microbiol Mol Biol Rev 65: 187–207.
Kim Y-H, Lee S-Y . (2013). Prevalence of putative periodontopahogen TM7 and dialister in dental plaque of Koreans. Int J Oral Biol 38: 55–59.
Kindt R, Coe R . (2005) Tree Diversity Analysis. A Manual and Software for Common Statistical Methods for Ecological and Biodiversity Studies. World Agroforestry Centre (ICRAF): Nairobi, Kenya, http://www.worldagroforestry.org/treesandmarkets/tree_diversity_analysis.asp.
Kitten T, Kinscherf TG, McEvoy JL, Willis DK . (1998). A newly identified regulator is required for virulence and toxin production in Pseudomonas syringae. Mol Microbiol 28: 917–929.
Kolenbrander PE . (2000). Oral microbial communities: biofilms, interactions, and genetic systems. Ann Rev Microbiol 54: 413–437.
Kolenbrander PE, Andersen RN, Blehert DS, Egland PG, Foster JS, Palmer RJJ . (2002). Communication among oral bacteria. Microbiol Mol Biol Rev 66: 486–505.
Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ . (2003). New bacterial species associated with chronic periodontitis. J Dental Res 82: 338–344.
Larsen MH, Leisner JJ, Ingmer H . (2010). The chitinolytic activity of Listeria monocytogenes EGD is regulated by carbohydrates but also by the virulence regulator PrfA. Appl Environ Microbiol 76: 6470–6476.
Lecuit M, Ohayon H, Braun L, Mengaud J, Cossart P . (1997). Internalin of Listeria monocytogenes with an intact leucine-rich repeat region is sufficient to promote internalization. Infect Immun 65: 5309–5319.
Legendre P, Gallagher ED . (2001). Ecologically meaningful transformations for ordination of species data. Oecologia 129: 271–280.
Lepp PW, Brinig MM, Ouverney CC, Palm K, Armitage GC, Relman DA . (2004). Methanogenic Archaea and human periodontal disease. Proc Natl Acad Sci USA 101: 6176–6181.
Liu B, Faller LL, Klitgord N, Mazumdar V, Ghodsi M, Sommer DD et al. (2012). Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS One 7: e37919.
Lux R, Miller JN, Park NH, Shi W . (2001). Motility and chemotaxis in tissue penetration of oral epithelial cell layers by Treponema denticola. Infect Immun 69: 6276–6283.
Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, Martin HG et al. (2007). Dissecting biological ‘dark matter’ with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci. USA 104: 11889–11894.
Marsh PD . (2006). Dental plaque as a biofilm and a microbial community-implications for health and disease. BMC Oral Health 6 (Suppl 1): S14.
McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR, Malmstrom RR et al. (2010). Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci USA 107: 16420–16427.
Mishima E, Sharma A . (2011). Tannerella forsythia invasion in oral epithelial cells requires phosphoinositide 3-kinase activation and clathrin-mediated endocytosis. Microbiology (Reading, Engl) 157: 2382–2391.
Murray DA, Wilton JMA . (2003). Lipopolysaccharide from the periodontal pathogen Porphyromonas gingivalis prevents apoptosis of HL60-derived neutrophils in vitro. Infect Immun 71: 7232–7235.
Nairz M, Schroll A, Sonnweber T, Weiss G . (2010). The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol 12: 1691–1702.
Ouverney CC, Armitage GC, Relman DA . (2003). Single-cell enumeration of an uncultivated TM7 subgroup in the human subgingival crevice. Appl Environ Microbiol 69: 6294–6298.
Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA et al. (2001). Bacterial diversity in human subgingival plaque. J Bacteriol 183: 3770–3783.
Peterson SN, Snesrud E, Liu J, Ong AC, Kilian M, Schork NJ et al. (2013). The dental plaque microbiome in health and disease. PLoS One 8: e58487.
Podar M, Abulencia CB, Walcher M, Hutchison D, Zengler K, Garcia JA et al. (2007). Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl Environ. Microbiol 73: 3205–3214.
Poroyko V, White JR, Wang M, Donovan S, Alverdy J, Liu DC et al. (2010). Gut Microbial Gene Expression in Mother-Fed and Formula-Fed Piglets. PLoS One 5: e12459.
Preshaw PM, Schifferle RE, Walters JD . (1999). Porphyromonas gingivalis lipopolysaccharide delays human polymorphonuclear leukocyte apoptosis in vitro. J Periodont Res 34: 197–202.
Quinlan AR, Hall IM . (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842.
Ramette A . (2007). Multivariate analyses in microbial ecology. FEMS Microbiol Ecol 62: 142–160.
Rams TE, Degener JE, van Winkelhoff AJ . (2012). Prevalence of β-lactamase-producing bacteria in human periodontitis. J Periodont Res 48: 493–499.
Relman DA . (2012). The human microbiome: ecosystem resilience and health. Nutr Rev 70 (Suppl 1): S2–S9.
Rhodes ER, Menke S, Shoemaker C, Tomaras AP, McGillivary G, Actis LA . (2007). Iron acquisition in the dental pathogen Actinobacillus actinomycetemcomitans: what does it use as a source and how does it get this essential metal? Biometals 20: 365–377.
Rodrigues PH, Progulske-Fox A . (2005). Gene expression profile analysis of Porphyromonas gingivalis during invasion of human coronary artery endothelial cells. Infect Immun 73: 6169–6173.
Rudney JD, Chen R, Sedgewick GJ . (2005). Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, and Tannerella forsythensis are components of a polymicrobial intracellular flora within human buccal cells. J Dent Res 84: 59–63.
Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D et al. (2012). Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol 13: R42.
Siqueira JF Jr, Rôças IN, Souto R, de Uzeda M, Colombo AP . (2000). Checkerboard DNA-DNA hybridization analysis of endodontic infections. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 89: 744–748.
Slots J . (2010). Human viruses in periodontitis. Periodontol 2000 53: 89–110.
Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RLJ . (1998). Microbial complexes in subgingival plaque. J Clin Periodontol 25: 134–144.
Soneson C, Delorenzi M . (2013). A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics 14: 91.
Supek F, Bošnjak M, Škunca N, Šmuc T . (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800.
Tang-Larsen J, Claesson R, Edlund MB, Carlsson J . (1995). Competition for peptides and amino acids among periodontal bacteria. J Periodont Res 30: 390–395.
Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A . (2011). Differential expression in RNA-seq: a matter of depth. Genome Res 21: 2213–2223.
Teles F, Haffajee AD, Socransky SS . (2007). Multiple displacement amplification as an aid in checkerboard DNA-DNA hybridization. Oral Microbiol Immunol 22: 118–125.
Thomas RZ, Zijnge V, Ciçek A, de Soet JJ, Harmsen HJM, Huysmans MCDNJM . (2012) Shifts in the microbial population in relation to in situ caries progression. Caries Res 46: 427–431.
Van Winkelhoff AJ, Winkel EG, Barendregt D, Dellemijn-Kippuw N, Stijne A, van der Velden U . (1997). beta-Lactamase producing bacteria in adult periodontitis. J Clin Periodontol 24: 538–543.
Wang C-H, Lin C-Y, Luo Y-H, Tsai P-J, Lin Y-S, Lin MT et al. (2005). Effects of oligopeptide permease in group a streptococcal infection. Infect Immun 73: 2881–2890.
Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL et al. (2013). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 42: 581–591.
Wright HJ, Chapple ILC, Matthews JB, Cooper PR . (2011). Fusobacterium nucleatum regulation of neutrophil transcription. J Periodont Res 46: 1–12.
Xia Q, Wang T, Taub F, Park Y, Capestany CA, Lamont RJ et al. (2007). Quantitative proteomics of intracellular Porphyromonas gingivalis. Proteomics 7: 4323–4337.
Ximenez-Fyvie LA, Haffajee AD, Socransky SS . (2000). Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J Clin Periodontol 27: 648–657.
Xiong X, Frank DN, Robertson CE, Hung SS, Markle J, Canty AJ et al. (2012). Generation and Analysis of a Mouse Intestinal Metatranscriptome through Illumina Based RNA-Sequencing. PLoS One 7: e36009.
Young MD, Wakefield MJ, Smyth GK, Oshlack A . (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11: R14.
Yuan L, Rodrigues PH, Bélanger M, Dunn W Jr, Progulske-Fox A . (2007). The Porphyromonas gingivalis clpB gene is involved in cellular invasion in vitro and virulence in vivo. FEMS Immunol Med Microbiol 51: 388–398.
Acknowledgements
Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health under award numbers DE021553 and DE021127. We were also grateful to Dr Mary-Ellen Davey for reviewing the manuscript and her useful comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Duran-Pinedo, A., Chen, T., Teles, R. et al. Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J 8, 1659–1672 (2014). https://doi.org/10.1038/ismej.2014.23
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.23
Keywords
This article is cited by
-
Lung microbiome: new insights into the pathogenesis of respiratory diseases
Signal Transduction and Targeted Therapy (2024)
-
Microbiota in health and diseases
Signal Transduction and Targeted Therapy (2022)
-
Long-term dynamics of the human oral microbiome during clinical disease progression
BMC Biology (2021)
-
Comparison of the modulatory effects of three structurally similar potential prebiotic substrates on an in vitro multi-species oral biofilm
Scientific Reports (2021)
-
Periodontitis associates with species-specific gene expression of the oral microbiota
npj Biofilms and Microbiomes (2021)
