
Abstract
‘Candidatus Microthrix parvicella’ is a lipid-accumulating, filamentous bacterium so far found only in activated sludge wastewater treatment plants, where it is a common causative agent of sludge separation problems. Despite attracting considerable interest, its detailed physiology is still unclear. In this study, the genome of the RN1 strain was sequenced and annotated, which facilitated the construction of a theoretical metabolic model based on available in situ and axenic experimental data. This model proposes that under anaerobic conditions, this organism accumulates preferentially long-chain fatty acids as triacylglycerols. Utilisation of trehalose and/or polyphosphate stores or partial oxidation of long-chain fatty acids may supply the energy required for anaerobic lipid uptake and storage. Comparing the genome sequence of this isolate with metagenomes from two full-scale wastewater treatment plants with enhanced biological phosphorus removal reveals high similarity, with few metabolic differences between the axenic and the dominant community ‘Ca. M. parvicella’ strains. Hence, the metabolic model presented in this paper could be considered generally applicable to strains in full-scale treatment systems. The genomic information obtained here will provide the basis for future research into in situ gene expression and regulation. Such information will give substantial insight into the ecophysiology of this unusual and biotechnologically important filamentous bacterium.
Similar content being viewed by others

Introduction
‘Candidatus Microthrix parvicella’ (henceforth called M. parvicella) is often found in activated sludge wastewater treatment plants, where it is commonly associated with the sludge separation problems of ‘bulking’ and ‘foaming’ (Eikelboom and van Buijsen, 1983; Jenkins et al., 1993; Rossetti et al., 2005; Wanner et al., 2010). Surveys of bulking/foaming municipal plants globally have often identified high abundances of M. parvicella as their most frequent cause (Wanner et al., 2010). Consequently, considerable effort has been directed at understanding its ecophysiology, hoping that this work would result in developing control strategies for it. A critical review of the data from these studies is given by Rossetti et al. (2005).
M. parvicella was originally defined by its distinct morphological features of an unsheathed, unbranched Gram-positive filament (van Veen, 1973; Eikelboom and van Buijsen, 1983). It has since been identified phylogenetically as a novel member of the Actinobacteria based on its 16S rRNA gene sequence (Blackall et al., 1994). Despite its repeated successful isolation into pure culture (van Veen, 1973; Eikelboom, 1975; Slijkhuis, 1983; Blackall et al., 1994; Seviour et al., 1994; Rossetti et al., 1997; Levantesi et al., 2006), its slow growth rate and an apparent recalcitrance to maintenance in pure culture has meant that no comprehensive characterisation has been achievable. Thus, it still has ‘Candidatus’ status (Blackall et al., 1994; Rossetti et al., 2005). However, some physiological information has been generated for the ‘Dutch isolate’ of Slijkhuis (1983) and the ‘Italian isolate RN1’ of Rossetti et al. (1997), while 16S rRNA gene sequence data is available only for the latter (Rossetti et al., 2005).
A closely related species ‘Candidatus Microthrix calida’, also isolated from industrial activated sludge, is the only other member of this genus. These two species differ only slightly in their trichome diameter, while sharing 95.7–96.7% sequence homology in their 16S rRNA gene (Levantesi et al., 2006). Fluorescence in situ hybridisation probes targeting the 16S rRNA of both have been described for their in situ identification and their application appears to indicate additional as yet undocumented phylogenetic diversity among organisms with an ‘M. parvicella’ morphotype (Erhart et al., 1997; Levantesi et al., 2006). Therefore, earlier morphology-based studies on the ‘M. parvicella’, where such unequivocal phylogenetic identity has not been determined, should be treated with caution.
Despite considerable research effort, only basic information is available on why M. parvicella might thrive in activated sludge plants. Full-scale plant studies indicate that this organism is physiologically highly versatile and able to adapt to marked changes in operational conditions (Rossetti et al., 2005), while the in situ and pure culture data are often contradictory (Andreasen and Nielsen, 1997, 1998; Tandoi et al., 1998). What is established is that in pure culture it is chemo-organoheterotrophic with a slow growth rate, and can tolerate hypoxic and cold conditions (Slijkhuis, 1983; Tandoi et al., 1998). These attributes might explain its reported preference for systems with long sludge retention times, low dissolved oxygen levels and those in colder climates (Rossetti et al., 2005). In situ carbon uptake studies indicate that it has a strong preference for lipid substrates, while pure culture and in situ studies also indicate a hydrophobic surface with associated exocellular lipase activity likely to assist in this (Tandoi et al., 1998; Nielsen et al., 2002). Its ability to compete for carbon substrates under unbalanced growth conditions makes it well suited to the alternating anoxic/oxic conditions of biological nutrient removal plants (Andreasen and Nielsen, 1997, 1998; Hesselsoe et al., 2005). Yet, other equally important aspects of its metabolism remain unclear. For example, a role for its observed polyphosphate storage and the source of energy used for lipid uptake and storage are yet to be explained. Obtaining a more thorough understanding of its metabolic potential should lead to better control strategies for M. parvicella in activated sludge. Any increase in understanding lipid accumulation mechanisms in bacteria will also be beneficial, as these have the potential for commercial application and are implicated in pathogen virulence (Alvarez and Steinbüchel, 2002; Kalscheuer, 2010).
In this study, we sequenced and annotated the genome of M. parvicella strain RN1 (Rossetti et al., 1997) and developed a metabolic model. A similar approach has enhanced our understanding of other activated sludge populations, including ‘Candidatus Accumulibacter phosphatis’ (henceforth called Accumulibacter) (García-Martín et al., 2006; He and McMahon, 2011) and Tetrasphaera spp., both polyphosphate-accumulating organisms (PAO) (Kristiansen et al., 2013), and the nitrite-oxidising ‘Candidatus Nitrospira defluvii’ (Lücker et al., 2010). In addition, we compared the obtained genome to two metagenomic data sets from Danish wastewater treatment plants (WWTPs) (Albertsen et al., 2012) to assess how relevant data from this pure culture is to the dominant strains present in full-scale WWTP communities. Combined with previous in situ study data this information will provide a comprehensive picture of the metabolic capability and potential of M. parvicella and provide a basis for future research into its in situ gene expression and regulation.
Materials and methods
Strain and culture conditions
M. parvicella strain RN1, originally isolated from the Rome North WWTP (Rossetti et al., 1997) (see Supplementary Figure S1 for phylogenetic placement), was grown for 4–5 weeks on freshly prepared R2A agar (Reasoner and Geldreich, 1985) at 20 °C and pH 7.2.
DNA extraction, sequencing library preparation and sequencing
DNA was isolated from colonies using the Fast DNA Spin Kit for Soil (MP Biomedicals, Solon, OH, USA) according to the manufacturer’s instructions. DNA concentration was measured with the dsDNA BR Assay Kit on a Qubit 2.0 Fluorometer (Invitrogen, Hellerup, Denmark) and its integrity verified after gel electrophoresis.
A DNA library from RN1 was prepared from 50 ng of DNA using the Nextera Kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s instructions, except that the ‘Clean-Up of Tagmented DNA’ step was conducted using the MinElute PCR Purification Kit (Qiagen, AB, Sollentuna, Sweden). Libraries were pair end (PE) sequenced (2 × 150 bp) using Truseq PE Cluster Kit v.3 (Illumina Inc.) and the TruSeq SBS Kit v.3-HS Sequencing Kit (Illumina Inc.) on an Illumina HiSeq 2000 (Illumina Inc.).
Genome assembly
Sequenced PE reads were imported into CLC genomics workbench v.5.1 (CLC Bio, Aarhus, Denmark) and quality trimmed using a minimum phred score of 20 and a minimum read length of 50, allowing no ambiguous nucleotides and removing Illumina sequencing adapters if found. Trimmed PE reads were assembled using CLC’s de novo assembly algorithm with a kmer of 63 and a minimum scaffold size of 500 bp. The assembly was evaluated by mapping PE reads to the assembled scaffolds using CLC’s map reads to reference function at 95% similarity. The mapping was exported in SAM format and Cytoscapeviz.pl (https://github.com/MadsAlbertsen/multi-metagenome/tree/master/cytoscapeviz) was used to track PE reads mapping to different scaffolds, and to calculate coverage of individual scaffolds. Subsequent visualisation using Cytoscape v.2.8.1 (Smoot et al., 2011) allowed manual refinement of the assembly. Completeness and potential contamination were evaluated by assessing 107 ‘essential single copy genes’ found in 95% of all sequenced bacteria (Dupont et al., 2012). This annotated sequence data has been submitted to DDBJ/EMBL/GenBank databases under accession nos. CANL01000001–CANL01000087.
Genome annotation and metabolic reconstruction
The contigs were uploaded to the ‘MicroScope’ annotation pipeline (Vallenet et al., 2006, 2009) and automatic annotations were validated manually for the genes involved in metabolic pathways of interest.
Comparison of the RN1 genome to full-scale community strains
Metagenome data sets were previously generated from biomass from two wastewater treatment plants with enhanced biological phosphorus removal (EBPR), Aalborg East (Albertsen et al., 2012) and Aalborg West (Albertsen et al., in preparation: Data set uploaded to MG-RAST (Meyer et al., 2008) ID: 4463936) WWTPs. Metagenome sequence reads were mapped to the genome of RN1 using CLC’s map reads to reference function, and requiring at least 70% similarity and a minimum read length of 50 bp. A three-dimensional recruitment plot was constructed, as described in Albertsen et al. (2012), to investigate the diversity among M. parvicella-related EBPR community strains. In addition, reads mapping at minimum 95% similarity were visualised using Circos (Krzywinski et al., 2009) to investigate differences between the RN1 genome and closely related community strains.
Results and Discussion
General features of the M. parvicella genome
The genome of the M. parvicella RN1 strain that was isolated from a conventional non-EBPR WWTP was sequenced and partially assembled. The RN1 genome assembly is 4.2 Mbp divided into 87 contigs owing to repeat regions that could not be resolved using the PE insert length used in this study (Table 1 and Supplementary Figure S2). The presence of 106 of the 107 selected ‘single copy essential genes’ (Dupont et al., 2012) suggests that the genome is essentially complete (Supplementary Table S1). According to the integrated microbial genome (IMG) database (Markowitz et al., 2012), the only absent gene at least also appears absent from genomes of some other organisms. Likewise, the three genes present in duplicate are also found in multiple copies in some genomes.
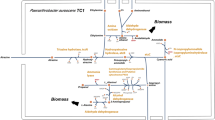
Analysis of the genome revealed that more than 40% of the predicted coding sequences (CDS) had no homologues to those found in other organisms, reflecting the lack of sequence information available for related organisms and Gram-positive bacteria in general. In fact, the genome of the relatively distantly related Acidimicrobium ferrooxidans (see Supplementary Figure S1 for phylogeny) is the sole representative of the Acidimicrobidae actinobacterial subclass, of which M. parvicella is a member. Obtaining genome information for such under-represented phylogenetic groups is of additional interest, but also makes annotation of the selected pathways in the RN1 genome difficult, with relatively low levels of supporting evidence for some genes. The abilities of M. parvicella to perform the metabolic pathways discussed here are based on the presence/absence of the majority of the genes required, with an emphasis on those considered unique to the pathway under consideration. This approach was coupled with the experimental data generated in earlier in situ and pure culture studies. A summary of the automated annotation of genes into clustered orthologous groups is given in Supplementary Table S2. A list of the curated annotated genes pertinent to selected pathways is given in Supplementary Table S3, and a metabolic overview is presented in Figure 1.

Diagrammatic representation of the proposed metabolic model for M. parvicella in EBPR-activated sludge treatment plants (a) in the absence of electron acceptors (anaerobic) and (b) in the presence of the electron acceptors oxygen (aerobic) or nitrate. Underlined products indicate putative storage or end products of metabolism. For more details on the pathways please refer to Supplementary Figure S3 in conjunction with Supplementary Table S3.
Comparison of central pathways in M. parvicella
Extrapolation of pure culture data to in situ systems is problematic and raises the question of how closely related and hence relevant such findings are to the ‘real world’. This concern is especially relevant to M. parvicella, where contradictions between pure culture and in situ physiological data are reported. The inherent difficulty in omic-based in situ studies in microbial ecology reflects largely the lack of reference genomes closely related to the strains of interest (Albertsen et al., 2012). Comparing the RN1 genome with the two metagenomes from full-scale Danish EBPR plants suggests that this non-EBPR system isolate is remarkably similar to those found in these full-scale EBPR communities (Figure 2). The similarity is much greater than that found for the Accumulibacter genome (García-Martín et al., 2006) when compared with the full-scale community strains in the same two metagenomes (Albertsen et al., 2012, in preparation). While some strain genomic variations do occur (discussed later), the key genes in the metabolic pathways discussed in this study occur in genomes of the M. parvicella pure culture and both dominant full-scale plant populations. Thus, although the discussions focus on the RN1 genome, its features and subsequent proposed metabolic model could be considered relevant generally to M. parvicella. During the review process of this manuscript, a draft genome sequence of the M. parvicella strain Bio17-1 was published (Muller et al., 2012). Genome sequence comparison of the RN1 and Bio17-1 was made using the JSpecies software (Richter and Rosselló-Móra, 2009), giving a calculated average nucleotide identity of 99% over the 89% of the genomes aligned by the programme. This supports the suggested low genomic diversity between strains of M. parvicella.

Graphical overview of the mapping of the short metagenome reads, from (a) Aalborg East and (b) Aalborg West WWTP, to the genome of M. parvicella RN1 as a function of per cent identity of each read. rRNA and tRNA genes have been masked. At the back of the figure, the summed coverage for a given genome position is shown.
Carbon source preference
In situ microautoradiography-fluorescence in situ hybridisation experiments have shown that M. parvicella has a strong preference for lipids as its carbon source, readily assimilating long-chain fatty acids (LCFAs), but not volatile fatty acids or simple sugars (Andreasen and Nielsen, 1997, 1998). The broader substrate uptake profiles reported for strain RN1 pure cultures (Tandoi et al., 1998; Tomei et al., 1999) may reflect that organisms become more specialized feeders in highly competitive natural environments than in axenic culture (Nielsen et al., 2010) (see Supplementary Text).
The M. parvicella RN1 genome contains putative genes for the β-oxidation of LCFAs to generate acetyl-CoA. Identification of putative exocellular triacylglycerol (TAG) lipase encoding genes for hydrolysis of lipids to their constituent LCFA and glycerol moieties is consistent with the exoenzyme lipase activity demonstrated in situ (Nielsen et al., 2002; Rossetti et al., 2005; Schade and Lemmer, 2005). A putative transporter gene for glycerol was annotated, although transport mechanism/s for LCFAs in Gram-positive bacteria are generally unclear (see Supplementary Text). The rapid uptake rate and storage of LCFA suggests that active transport is involved (Andreasen and Nielsen, 2000). Glycerol, released after lipid hydrolysis, reportedly is not utilised by strain RN1 as a sole carbon source (Tomei et al., 1999). However, it is assimilated in situ, but only in the presence of LCFA (Kindaichi et al., 2012), consistent with TAGs acting as major carbon storage compounds (see later).
Central intermediary metabolism
The RN1 genome contains candidate genes for the pentose phosphate pathway and the Embden–Meyerhof–Parnas glycolysis pathway, while key genes in the Entner–Doudoroff (ED) version of this pathway are absent. All candidate genes for the complete oxidative tricarboxylic acid (TCA) cycle were also located. The genome also contains putative genes for two suggested bypasses of the α-oxoglutarate dehydrogenase complex in the TCA cycle, providing metabolic flexibility. One is a ferredoxin-dependent 2-oxoglutarate oxidoreductase (EC 1.2.7.3) reducing 2-oxoglutarate to succinyl-CoA, and is associated with microaerophilic and anaerobic lifestyles in other bacteria (Mai and Adams, 1996; Hughes et al., 1998; Huynen et al., 1999). Its presence may explain in part the microaerophilic nature of M. parvicella (Slijkhuis and Deinema, 1988; Tandoi et al., 1998; Andreasen and Nielsen, 2000). The other bypass is the γ-aminobutyric acid shunt (Rosenqvist et al., 1973). Accumulation of γ-aminobutyric acid is reported for activated sludge biomass with glutamate as supplied carbon source, where it may be utilised as an energy storage compound (Satoh et al., 1998). The reported correlation between high ammonia levels and high M. parvicella abundances in wastewater treatment plants (Kruit et al., 2002; Tsai et al., 2003) might be explained if the former serves as a precursor for the production of γ-aminobutyric acid, which functions as a carbon and nitrogen storage compound, and/or as an intermediate in an alternative TCA cycle (see Figure 1). Genes encoding enzymes of the glyoxylate shunt allowing growth on acetyl-CoA and generating compounds, including LCFAs (Kornberg and Krebs, 1957), are absent from the M. parvicella genome. Instead, it appears to utilise the anaplerotic ethylmalonyl-CoA pathway (Erb et al., 2007, 2009).
Carbon storage material
Although unable to utilise other carbon sources anaerobically, M. parvicella has a large storage capacity (Tandoi et al., 1998; Hesselsoe et al., 2005). Electron microscope images indicate large electron transparent storage inclusions, thought to be neutral lipids (Slijkhuis, 1983; Tandoi et al., 1998; Nielsen et al., 2002). In situ and pure culture studies suggest that LCFAs are stored in their esterified form (Slijkhuis et al., 1984; Nielsen et al., 2002; Rossetti et al., 2005). This is confirmed with the genome suggesting an ability to store TAG, a triacyl ester of glycerol, and a common feature of the Actinobacteria (Alvarez and Steinbüchel, 2002). Whether M. parvicella can store polyhydroxylalkanoates is unclear from the genomic analyses, and is discussed elsewhere (see Supplementary Text). The atfA is the key gene in TAG synthesis and codes for a bifunctional enzyme having waxy ester synthesis and acyl-CoA:DAG acyltransferase activities (Kalscheuer and Steinbüchel, 2003). The RN1 genome appears to possess at least 10 putative copies of this gene, thus falling within the numerical range for other oleaginous Actinobacteria (Kalscheuer, 2010). The encoded enzyme appears to have low substrate specificity, incorporating structurally diverse acyl groups, indicating that a diverse range of storage TAGs can be synthesised. In addition to its role in providing an energy source, TAG storage may also provide an advantage in mixed liquors with high lipid contents, by neutralising potentially toxic levels of LCFAs (Garton et al., 2002), including the selective incorporation of those LCFAs able to disrupt membrane fluidity (Murphy, 1993; Alvarez and Steinbüchel, 2002).
Studies with Mycobacterium tuberculosis have shown that genes involved in TAG synthesis are upregulated at low oxygen tensions (Daniel et al., 2004). A similar feature was observed in Rhodococcus spp., where TAGs were considered to act as electron sinks to cope with electron acceptor-limiting conditions (Alvarez, 2006). This may help explain why M. parvicella thrives under low oxygen conditions.
With Accumulibacter PAO, anaerobic hydrolysis of aerobically synthesised glycogen stores, followed by glycolysis and flexible operation of the TCA cycle supply some of the energy and reducing equivalents for carbon uptake and storage (He and McMahon, 2011). A similar strategy was suggested for M. parvicella (Nielsen et al., 2002), but genes encoding the key enzymes for glycogen metabolism are absent. One possible alternative energy source is trehalose, potentially serving a similar function to glycogen in Accumulibacter, and genes for its synthesis and hydrolysis were found. No transporter genes for trehalose were identified, and RN1 cannot utilise it as its sole carbon source (Tomei et al., 1999), suggesting that exogenous sources are not utilisable. The polyphosphate-accumulating activated sludge isolate Microlunatus phosphovorus also accumulates trehalose (Santos et al., 1999). However, no anaerobic:aerobic cycling of trehalose has been observed and no role elucidated. Trehalose synthesis may serve potentially several alternative purposes for M. parvicella as proposed for other organisms. These include imparting resistance to desiccation, heat, oxidative and osmotic stresses, and being incorporated into cell walls of some Gram-positive bacteria (Elbein et al., 2003). Trehalose accumulation can increase survival of E. coli at low temperatures (4 °C) (Kandror et al., 2002), and may serve a similar function with M. parvicella. It can tolerate low temperature axenically (Slijkhuis, 1983; Tandoi et al., 1998) and increases in abundance in full-scale plants in colder climates and seasons (Seviour et al., 1990; Kristensen et al., 1994; Bejvl et al., 2004; Mielczarek et al., 2012).
Phosphorus metabolism
The role of the observed polyphosphate storage in M. parvicella is unclear, but its synthesis has been suggested to be in response to cell stress (Erhart et al., 1997; Tandoi et al., 1998). Of particular interest is whether or not the polymer serves as an anaerobic energy reserve for carbon uptake, as has been proposed for the Accumulibacter and Tetrasphaera PAO (Kong et al., 2005; Saunders et al., 2007; Kristiansen et al., 2013). However, in situ studies show no substantive aerobic:anaerobic cycling of polyphosphate linked to carbon uptake for the M. parvicella, suggesting a different role in its metabolism (Andreasen and Nielsen, 2000). All the putative genes necessary for the synthesis and hydrolysis of polyphosphate were located in the RN1 genome (see Supplementary Text). This theoretically permits the generation of ATP from direct phosphorylation of AMP, catalyzed by the combined activities of the polyphosphate and adenylate kinases (Ishige and Noguchi, 2000). The presence of genes encoding a polyphosphate glucokinase (EC 2.7.1.63) and a polyphosphate/ATP NAD+ kinase (EC 2.7.1.23) might indicate a role for polyP in direct phosphorylation of intermediates involved in energy metabolism.
However, an important difference between the PAO and M. parvicella is in the phosphate transport systems encoded by their genomes. For RN1, the genes were located for the high-affinity Pst phosphorus uptake system, but absent for the low-affinity Pit system, while both are present in the Accumulibacter and Tetrasphaera PAO genomes (García-Martín et al., 2006; Kristiansen et al., 2013). This suggests that phosphorus uptake for M. parvicella may occur only under phosphorus-limiting conditions (Kornberg et al., 1999). An additional role of the Pit system in the PAO is the proton motive force generated from the anaerobic efflux of phosphorus with the symport of a proton, which is suggested to energise carbon uptake (Saunders et al., 2007; Burow et al., 2008). Thus, polyphosphate storage does not appear to have the same central role in the energy metabolism of M. parvicella. However, this does not preclude the possibility of a reduced reliance on the polymer as an energy source, and concomitant lower levels of its cyclical transformations. These changes may be below the detection limits of the fluorescence in situ hybridisation-microautoradiography method applied in the study of Andreasen and Nielsen (2000).
Nitrogen metabolism
Axenically strain RN1 reduces nitrate to nitrite, but does not grow under anaerobic conditions with or without the presence of nitrate (Tandoi et al., 1998; Rossetti et al., 2002). In situ microautoradiography studies show that the level of metabolic activity, based on 14CO2 assimilation, of M. parvicella is similar in the presence of either oxygen or nitrate (Andreasen and Nielsen, 2000; Hesselsoe et al., 2005). These observations are supported by the location of putative genes for a nitrate transporter and a nitrate reductase complex (EC 1.7.99.4).
Pure culture studies indicate that reduction of nitrate by M. parvicella does not proceed past nitrite (Tandoi et al., 1998; Hesselsoe et al., 2005). Yet, a putative nirK gene coding for the copper-containing nitrite reductase (EC 1.7.2.1) was located in the RN1 genome. Preliminary details from the annotation of the M. parvicella str. Bio17-1 genome suggest the absence of a nitrite reductase (Muller et al., 2012). However, a BLAST search of this genome with the putative gene from RN1 identified a CDS with 99.9% nucleotide sequence identity. Genes coding for enzymes capable of denitrification beyond nitric oxide were not identified in the RN1 genome, although others involved in nitric oxide detoxification were (see Supplementary Text). In situ fluorescence in situ hybridisation-microautoradiography experiments indicate that M. parvicella tolerates anaerobic starvation for longer in the presence of nitrite, suggesting that this filament benefits somehow from its presence (Andreasen and Nielsen, 2000). In situ data showed considerably less 14CO2 incorporation into the biomass with nitrite than with nitrate as an electron acceptor. Thus, substantial anaerobic activity was not supported by the former (Hesselsoe et al., 2005), but was still above background levels, which would indicate some nitrite reduction. Its extent was restricted possibly by nitric oxide accumulation or other environmental conditions (Zumft, 1997).
A metabolic model for M. parvicella in activated sludge systems
On the basis of these genome annotations of key pathways and the findings of pure culture and in situ studies, a metabolic model for M. parvicella is presented here (see Figure 1). It focuses on its carbon, nitrogen and phosphorus metabolism pertaining to its survival under the dynamic conditions of EBPR-activated sludge systems, where biomass is cycled repeatedly through carbon-rich anaerobic and carbon-deficient aerobic conditions (Seviour et al., 2003). However, most of the following discussion is equally relevant to other system configurations where this organism is also abundant.
Anaerobic transformations
All published in situ data indicate that this organism prefers lipids as growth substrates (Andreasen and Nielsen, 1997, 1998; Nielsen et al., 2002). The postulated hydrophobic cell wall facilitates filament attachment to lipids, and exocellular lipases hydrolyze these to generate LCFA that are then taken up by as yet unknown mechanisms. Once transported, these are activated to acyl-CoAs, which are attached sequentially to glycerol-3P backbones to synthesise the TAGs used for carbon storage. Given the high uptake and storage rates recorded for these LCFAs, active transport mechanisms are probably involved. Therefore, the source of energy for this uptake and storage needs consideration. As they also have the ability to assimilate LCFA in the absence of any exogenous glycerol supply (Andreasen and Nielsen, 1997), the carbon source used and redox balance conditions also require thought.
The carbon skeletons for synthesising the glycerol-3P backbone of TAG may arise from partial oxidation of LCFA by the β-oxidation pathway, thus releasing acetyl-CoA that is then converted to glycerol involving the ethylmalonyl-CoA and gluconeogenesis pathways, and/or from catabolism of intracellular trehalose deposits (Figure 1). Any additional NAD(P)H requirement may be met from partial operation of the TCA cycle. Whether M. parvicella operates a functional anaerobic TCA cycle beyond the production of succinate is unclear. In Tetrasphaera elongata, glucose is fermented to succinate, which is then exported from the cell (Kristiansen et al., 2013). In Accumulibacter several variations of the TCA cycle probably operate under anaerobic conditions to regenerate oxidised quinones, including novel cytochrome and fumarate reductase activities, required for succinate dehydrogenase function (García-Martín et al., 2006; He and McMahon, 2011). It is also possible that fatty acid β-oxidation provides the required reducing equivalents anaerobically. Potential reverse operation of a putative acetyl-CoA synthetase (EC 6.2.1.13) on the subsequent acetyl-CoA and possibly propionyl-CoA produced from the branched ethylmalonyl-CoA pathway may generate the ATP for LCFA uptake and activation, with consequent production of acetate and propionate as by-products. Additional ATP may also be supplied by the hydrolysis of polyphosphate stores. Even though substantial polyphosphate cycling is not observed, ATP generated from its hydrolysis may only partially contribute to the energy required. In addition, the anaerobic energy demand is likely to be markedly reduced, given that catabolism of each LCFA molecule generates substantially more energy than that generated from the simple sugars and volatile fatty acids considered as energy sources in the metabolic models for Tetrasphaera (Kristiansen et al., 2013) and Accumulibacter (García-Martín et al., 2006).
Aerobic transformations
Under aerobic conditions, the stored TAGs appear to be processed using the β-oxidation and ethylmalonyl-CoA pathways into the TCA cycle, whose operation would provide the energy and intermediates required for M. parvicella growth. This storage compound is functionally analogous to the fate of PHA in Accumulibacter. Such transformations for TAGs are supported at least partly by in situ studies where radiolabelled oleate was tracked into neutral lipids anaerobically, and under subsequent aerobic conditions CO2 was released and polar membrane lipids formed (Nielsen et al., 2002). If trehalose reserves are utilised anaerobically, then their replenishment would thus occur aerobically. The trigger for such storage may be oxidative stress, as Candida albicans increases its intracellular trehalose levels in response to such conditions (Alvarez-Peral et al., 2002). The genome sequence data also suggest that nitrate and nitrite may substitute for oxygen as the terminal electron acceptor for anaerobic respiration, although only nitrate reduction ability is supported by experimental evidence (Tandoi et al., 1998; Hesselsoe et al., 2005).
Genomic variations between M. parvicella strains
Comparisons of the RN1 genome with the two metagenomes from full-scale EBPR plants indicate some genetic variation between strains. These are represented in Figure 3. Most regions include coding regions for which no function could be assigned because of the lack of homology to existing sequences in the database. Many of the identifiable genes are associated with mobile genetic elements or with cell envelope and exopolysaccharide synthesis. Intraspecies variation in exopolysaccharide gene cassettes has been reported for Accumulibacter, the suggestion being that these relate to an adaptive defence to viral predation, given that phage genomes can encode strain-specific polysaccharases (Kunin et al., 2008; Albertsen et al., 2012). Such differences in polysaccharide structures may influence cell hydrophobicity, in which case M. parvicella strains may differ in their abilities to stabilise foams in activated sludge.

Circular visualisation of coverage of the M. parvicella RN1 genome in the metagenomes prepared from Aalborg East and Aalborg West WWTPs. The inner full circle represents the RN1 genome, where lines indicate contig breaks. rRNA and tRNA genes have been masked. The outer circles represent metagenome coverage. The inner sections represent selected regions in the RN1 genome that are either under-represented or missing from the metagnomes. Annotation details for these sections are given in Supplementary Table S4.
Identified functional differences between the RN1 strain and related EBPR community strains include the presence of genes involved in mercury resistance and fructose utilisation (Figure 3). Despite the presence of the latter genes in the RN1 genome, axenic studies indicate an inability to utilise fructose, at least as sole carbon source (Tomei et al., 1999). However, the corresponding expressed enzymes may permit utilisation of fructose as a precursor for the synthesis of other exopolysaccharides or cellular structural components. Thus, identifiable differences generally appear to relate to defence against environmental challenges to the organism, giving stability to M. parvicella populations and enhancing their ability to survive under the rapidly fluctuating conditions of full-scale treatment plants.
Concluding remarks
The metabolic model presented in this study represents the first insight into the metabolic potential of the M. parvicella. The key to its survival in activated sludge plants appears to be its ability to cycle TAG polymers as an internal supply of carbon and energy. Metagenome comparisons indicate that the RN1 strain is remarkably similar to the abundant in situ strains, where little genetic diversity is evident. This indicates that this model should be considered relevant to the important environmental strains present in full-scale activated sludge processes. Such information represents a valuable step forward in understanding the ecology of this biotechnologically important organism, and importantly provides the foundation for subsequent metagenomic, metatranscriptomic and metaproteomic studies that will ultimately clarify the details of their metabolism.
Accession codes
References
Albertsen M, Hansen LB, Saunders AM, Nielsen PH, Nielsen KL . (2012). A metagenome of a full-scale microbial community carrying out enhanced biological phosphorus removal. ISME J 6: 1094–1106.
Alvarez HM . (2006). Bacterial triacylglycerols. In: Triglycerides and Cholesterol Research. Welson LT (eds) Nova Science Publishers: NewYork, NY, USA, pp 159–176.
Alvarez HM, Steinbüchel A . (2002). Triacylglycerols in prokaryotic microorganisms. Appl Microbiol Biotechnol 60: 367–376.
Alvarez-Peral FJ, Zaragoza O, Pedreno Y, Arguelles JC . (2002). Protective role of trehalose during severe oxidative stress caused by hydrogen peroxide and the adaptive oxidative stress response in Candida albicans. Microbiology 148: 2599–2606.
Andreasen K, Nielsen PH . (1997). Application of microautoradiography to the study of substrate uptake by filamentous microorganisms in activated sludge. Appl Environ Microbiol 63: 3662–3668.
Andreasen K, Nielsen PH . (1998). In situcharacterization of substrate uptake by Microthrix parvicella using microautoradiography. Water Sci Technol 37 ((4-5)): 19–26.
Andreasen K, Nielsen PH . (2000). Growth of Microthrix parvicella in nutrient removal activated sludge plants: studies of in situ physiology. Water Res 34: 1559–1569.
Bejvl Z, Matuska P, Stara J, Chudoba P . (2004). Performances of three R-AN-D-N wastewater treatment plants in the Czech Republic. Water Sci Technol 50 ((7)): 249–255.
Blackall LL, Seviour EM, Cunningham MA, Seviour RJ, Hugenholtz P . (1994). 'Microthrix parvicella’ is a novel, deep branching member of the actinomycetes subphylum. Syst Appl Microbiol 17: 513–518.
Burow L, Mabbett A, McEwan A, Bond P, Blackall L . (2008). Bioenergetic models for acetate and phosphate transport in bacteria important in enhanced biological phosphorus removal. Environ Microbiol 10: 87–98.
Daniel J, Deb C, Dubey VS, Sirakova TD, Abomoelak B, Morbidoni HR et al. (2004). Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol 186: 5017–5030.
Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R et al. (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199.
Eikelboom DH . (1975). Filamentous organisms observed in activated sludge. Water Res 9: 365–388.
Eikelboom DH, van Buijsen HJJ . (1983) Microscopic Sludge Investigation Manual. TNO Research Institute of Environmental Hygiene: Delft, Holland.
Elbein AD, Pan YT, Pastuszak I, Carroll D . (2003). New insights on trehalose: a multifunctional molecule. Glycobiology 13: 17R–27R.
Erb TJ, Berg IA, Brecht V, Muller M, Fuchs G, Alber BE . (2007). Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc Natl Acad Sci USA 104: 10631–10636.
Erb TJ, Fuchs G, Alber BE . (2009). (2S)-methylsuccinyl-CoA dehydrogenase closes the ethylmalonyl-CoA pathway for acetyl-CoA assimilation. Mol Microbiol 73: 992–1008.
Erhart R, Bradford D, Seviour RJ, Amann R, Blackall LL . (1997). Development and use of fluorescent in situ hybridization probes for the detection and identification of ‘Microthrix parvicella’ in activated sludge. Syst Appl Microbiol 20: 310–318.
García-Martín H, Ivanova N, V K, Warnecke F, Barry K, McHardy A et al. (2006). Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat Biotechnol 24: 1263–1269.
Garton NJ, Christensen H, Minnikin DE, Adegbola RA, Barer MR . (2002). Intracellular lipophilic inclusions of mycobacteria in vitro and in sputum. Microbiology 148: 2951–2958.
He S, McMahon KD . (2011). Microbiology of ‘Candidatus Accumulibacter’ in activated sludge. Microbial Biotechnol 4: 603–619.
Hesselsoe M, Nielsen JL, Roslev P, Nielsen PH . (2005). Isotope labeling and microautoradiography of active heterotrophic bacteria on the basis of assimilation of 14CO2 . Appl Environ Microbiol 71: 646–655.
Hughes NJ, Clayton CL, Chalk PA, Kelly DJ . (1998). Helicobacter pylori porCDAB and oorDABC genes encode distinct pyruvate:flavodoxin and 2-oxoglutarate:acceptor oxidoreductases which mediate electron transport to NADP. J Bacteriol 180: 1119–1128.
Huynen MA, Dandekar T, Bork P . (1999). Variation and evolution of the citric-acid cycle: a genomic perspective. Trends Microbiol 7: 281–291.
Ishige K, Noguchi T . (2000). Inorganic polyphosphate kinase and adenylate kinase participate in the polyphosphate:AMP phosphotransferase activity of Escherichia coli. Proc Natl Acad Sci USA 97: 14168–14171.
Jenkins D, Richard MC, Daigger GT . (1993) Manual on the Causes and Control of Activated Sludge Bulking and Foaming. Lewis Publishers: New York, NY, USA.
Kalscheuer R . (2010). Genetics of wax ester and triacylglycerol biosynthesis in Bacteria. In: Handbook of Hydrocarbon and Lipid Microbiology. Timmis KN (eds). pringer: Berlin, Heidelberg, Germany, pp 527–527-535.
Kalscheuer R, Steinbüchel A . (2003). A novel bifunctional wax ester synthase/acyl-CoA:diacylglycerol acyltransferase mediates wax ester and triacylglycerol biosynthesis in Acinetobacter calcoaceticus ADP1. J Biol Chem 278: 8075–8082.
Kandror O, DeLeon A, Goldberg AL . (2002). Trehalose synthesis is induced upon exposure of Escherichia coli to cold and is essential for viability at low temperatures. Proc Natl Acad Sci USA 99: 9727–9732.
Kindaichi T, Nierychlo M, Kragelund C, Nielsen JL, Nielsen PH . (2012). High and stable substrate specificities of microorganisms in enhanced biological phosphorus removal plants. Environ Microbiol doi:10.1111/1462-2920.12074 (in press).
Kong Y, Nielsen JL, Nielsen PH . (2005). Identity and ecophysiology of uncultured actinobacterial polyphosphate-accumulating organisms in full-scale enhanced biological phosphorus removal plants. Appl Environ Microbiol 71: 4076–4085.
Kornberg A, Rao NN, Ault-Riché D . (1999). Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem 68: 89–125.
Kornberg HL, Krebs HA . (1957). Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature 179: 988–991.
Kristensen GH, Jørgensen PE, Nielsen PH . (1994). Settling characteristics of activated sludge in Danish treatment plants with biological nutrient removal. Water Sci Technol 29 ((7)): 157–165.
Kristiansen R, Nguyen HTT, Saunders AM, Nielsen JL, Wimmer R, Le VQ et al. (2013). A metabolic model for members of the genus Tetrasphaera involved in enhanced biological phosphorus removal. ISME J 7: 543–554.
Kruit J, Hulsbeek J, Visser A . (2002). Bulking sludge solved?!. Water Sci Technol 46 ((1-2)): 457–464.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645.
Kunin V, He S, Warnecke F, Peterson SB, Garcia-Martin H, Haynes M et al. (2008). A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res 18: 293–297.
Levantesi C, Rossetti S, Thelen K, Kragelund C, Krooneman J, Eikelboom D et al. (2006). Phylogeny, physiology and distribution of 'Candidatus Microthrix calida', a new Microthrix species isolated from industrial activated sludge wastewater treatment plants. Environ Microbiol 8: 1552–1563.
Lücker S, Wagner M, Maixner F, Pelletier E, Koch H, Vacherie B et al. (2010). A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc Natl Acad Sci USA 107: 13479–13484.
Mai X, Adams MW . (1996). Characterization of a fourth type of 2-keto acid-oxidizing enzyme from a hyperthermophilic archaeon: 2-ketoglutarate ferredoxin oxidoreductase from Thermococcus litoralis. J Bacteriol 178: 5890–5896.
Markowitz VM, Chen IA, Palaniappan K, Chu K, Szeto E, Grechkin Y et al. (2012). IMG: the intergrated microbial genomes database and comparative analysis system. Nucleic Acids Res 40: D115–D122.
Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M et al. (2008). The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform 9: 386.
Mielczarek AT, Kragelund C, Eriksen PS, Nielsen PH . (2012). Population dynamics of filamentous bacteria in Danish wastewater treatment plants with nutrient removal. Water Res 46: 3781–3795.
Muller EE, Pinel N, Gillece JD, Schupp JM, Price LB, Engelthaler DM et al. (2012). Genome sequence of 'Candidatus Microthrix parvicella' Bio17-1, a long-chain-fatty-acid-accumulating filamentous actinobacterium from a biological wastewater treatment plant. J Bacteriol 194: 6670–6671.
Murphy DJ . (1993). Structure, function and biogenesis of storage lipid bodies and oleosins in plants. Prog Lipid Res 32: 247–280.
Nielsen PH, Mielczarek AT, Kragelund C, Nielsen JL, Saunders AM, Kong Y et al. (2010). A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res 44: 5070–5088.
Nielsen PH, Roslev P, Dueholm TE, Nielsen JL . (2002). Microthrix parvicella, a specialized lipid consumer in anaerobic-aerobic activated sludge plants. Water Sci Technol 46 ((1-2)): 73–80.
Reasoner DJ, Geldreich EE . (1985). A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol 49: 1–7.
Richter M, Rosselló-Móra R . (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106: 19126–19131.
Rosenqvist H, Kasula H, Reunanen O, Nurmikko V . (1973). The 4-aminobutyrate pathway and 2-oxoglutarate dehydrogenase in Escherichia coli. Acta Chem Scand 27: 3091–3100.
Rossetti S, Christensson C, Blackall LL, Tandoi V . (1997). Phenotypic and phylogenetic description of an Italian isolate of 'Microthrix parvicella'. J App Microbiol 82: 405–410.
Rossetti S, Tomei MC, Levantesi C, Ramadori R, Tandoi V . (2002). 'Microthrix parvicella': a new approach for kinetic and physiological characterization. Water Sci Technol 46 ((1-2)): 65–72.
Rossetti S, Tomei MC, Nielsen PH, Tandoi V . (2005). 'Microthrix parvicella', a filamentous bacterium causing bulking and foaming in activated sludge systems: a review of current knowledge. FEMS Microbiol. Rev 29: 49–64.
Santos MM, Lemos PC, Reis MA, Santos H . (1999). Glucose metabolism and kinetics of phosphorus removal by the fermentative bacterium Microlunatus phosphovorus. Appl Environ Microbiol 65: 3920–3928.
Satoh H, Mino T, Matsuo T . (1998). Anaerobic uptake of glutamate and aspartate by enhanced biological phosphorus removal activated sludge. Water Sci Technol 37 ((4-5)): 579–582.
Saunders A, Mabbett A, McEwan A, Blackall L . (2007). Proton motive force generation from stored polymers for the uptake of acetate under anaerobic conditions. FEMS Microbiol Lett 274: 245–251.
Schade M, Lemmer H . (2005). Lipase activities in activated sludge and scum—comparison of new and conventional techniques. Acta Hydrochim Hydrobiol 33: 210–215.
Seviour EM, Williams C, DeGrey B, Soddell JA, Seviour RJ, Lindrea KC . (1994). Studies on filamentous bacteria from Australian activated sludge plants. Water Res 28: 2335–2342.
Seviour EM, Williams CJ, Seviour RJ, Soddell JA, Lindrea KC . (1990). A survey of filamentous bacterial populations from foaming activated sludge plants in eastern states of Australia. Water Res 24: 493–498.
Seviour RJ, Mino T, Onuki M . (2003). The microbiology of biological phosphorus removal in activated sludge systems. FEMS Microbiol Rev 27: 99–127.
Slijkhuis H . (1983). Microthrix parvicella, a filamentous bacterium isolated from activated sludge: cultivation in a chemically defined medium. Appl Environ Microbiol 46: 832–839.
Slijkhuis H, Deinema MH . (1988). Effect of environmental conditions on the occurrence of Microthrix parvicella in activated sludge. Water Res 22: 825–828.
Slijkhuis H, van Groenestijn JW, Kylstra DJ . (1984). Microthrix parvicella, a filamentous bacterium from activated sludge: growth on Tween 80 as carbon and energy source. J Gen Microbiol 130: 2035–2042.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T . (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27: 431–432.
Tandoi V, Rossetti S, Blackall LL, Majone M . (1998). Some physiological properties of an italian isolate of 'Microthrix parvicella'. Water Sci Technol 37 ((1-2)): 1–8.
Tomei MC, Levantesi C, Rossetti S, Tandoi V . (1999). Microbiological characterisation of pure cultures and its relevance to modelling and control of bulking phenomena. Water Sci Technol 39 ((1)): 21–29.
Tsai MW, Wentzel MC, Ekama GA . (2003). The effect of residual ammonia concentration under aerobic conditions on the growth of Microthrix parvicella in biological nutrient removal plants. Water Res 37: 3009–3015.
Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A et al. (2009). MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford) 2009: bap021.
Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S et al. (2006). MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res 34: 53–65.
van Veen WL . (1973). Bacteriology of activated sludge, in particular the filamentous bacteria. Antonie van Leeuwenhoek 39: 189–205.
Wanner J, Kragelund C, Nielsen PH . (2010). Microbiology of bulking. In: Microbial Ecology of Activated Sludge. Seviour RJ, Nielsen PH (eds). IWA Publishing: London, UK, pp 191–214.
Zumft WG . (1997). Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61: 533–616.
Acknowledgements
This study was funded by Aalborg University and the Danish Research Council for Strategic Research via the Research Centre ‘EcoDesign-MBR’. The LABGeM team at Genoscope are also acknowledged for providing access to their automated annotation pipeline, and for database maintenance and technical assistance. We thank Dr Steve Petrovski for providing technical assistance and helpful discussion.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Jon McIlroy, S., Kristiansen, R., Albertsen, M. et al. Metabolic model for the filamentous ‘Candidatus Microthrix parvicella’ based on genomic and metagenomic analyses. ISME J 7, 1161–1172 (2013). https://doi.org/10.1038/ismej.2013.6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.6
Keywords
This article is cited by
-
Bacterial community and filamentous population of industrial wastewater treatment plants in Belgium
Applied Microbiology and Biotechnology (2024)
-
Prokaryotic diversity and potentially pathogenic bacteria in vended foods and environmental samples
Annals of Microbiology (2021)
-
Roles of bacteriophages, plasmids and CRISPR immunity in microbial community dynamics revealed using time-series integrated meta-omics
Nature Microbiology (2020)
-
Integration of time-series meta-omics data reveals how microbial ecosystems respond to disturbance
Nature Communications (2020)
-
Illumina sequencing for the identification of filamentous bulking and foaming bacteria in industrial activated sludge plants
International Journal of Environmental Science and Technology (2018)
