
Abstract
We investigated methanotrophic bacteria in slightly alkaline surface water (pH 7.4–8.7) of oilsands tailings ponds in Fort McMurray, Canada. These large lakes (up to 10 km2) contain water, silt, clay and residual hydrocarbons that are not recovered in oilsands mining. They are primarily anoxic and produce methane but have an aerobic surface layer. Aerobic methane oxidation was measured in the surface water at rates up to 152 nmol CH4 ml−1 water d−1. Microbial diversity was investigated via pyrotag sequencing of amplified 16S rRNA genes, as well as by analysis of methanotroph-specific pmoA genes using both pyrosequencing and microarray analysis. The predominantly detected methanotroph in surface waters at all sampling times was an uncultured species related to the gammaproteobacterial genus Methylocaldum, although a few other methanotrophs were also detected, including Methylomonas spp. Active species were identified via 13CH4 stable isotope probing (SIP) of DNA, combined with pyrotag sequencing and shotgun metagenomic sequencing of heavy 13C-DNA. The SIP-PCR results demonstrated that the Methylocaldum and Methylomonas spp. actively consumed methane in fresh tailings pond water. Metagenomic analysis of DNA from the heavy SIP fraction verified the PCR-based results and identified additional pmoA genes not detected via PCR. The metagenome indicated that the overall methylotrophic community possessed known pathways for formaldehyde oxidation, carbon fixation and detoxification of nitrogenous compounds but appeared to possess only particulate methane monooxygenase not soluble methane monooxygenase.
Similar content being viewed by others


Introduction
Canadian oilsands, primarily located in northern Alberta, represent one of the world’s largest petroleum reserves, estimated at 169 billion barrels (Energy Resources Conservation Board, 2012). Removing solids from the highly viscous oil requires extraction with 15–20 volumes of hot water per volume of oil extracted and subsequent treatment with chemical solvents. This process produces large amounts of tailings comprised of water, silt, clay, residual bitumen and small amounts of unrecovered solvents. Because the bitumen content of oilsands deposits is only about 10% by weight, the total volume ratio of waste tailings to oil produced is 12–14 (Kasperski and Mikula, 2011). Although new management options are being investigated, at present tailings are usually stored in open basins. As the silt and clay particles settle, water in the basins is recycled for reuse in the extraction procedure, so the net fresh water use in mining operations is only about three volumes of fresh water per volume of oil produced (Allen, 2008). Proper management of tailings ponds presents several environmental challenges, particularly limiting the contamination of surrounding aquifers with pollutants such as toxic metals, naphthenic acids and polycyclic aromatic compounds (Quagraine et al., 2005; Kelly et al., 2009, 2010) and limiting efflux of methane, a greenhouse gas with 25 times the global warming potential of CO2 per molecule (Solomon et al., 2007).
The tailings ponds in northern Alberta cover a total area of over 170 km2 (Government of Alberta, 2012). The large amounts of hydrocarbons in tailings, particularly residual bitumen, as well as reduced chemicals such as sulfides, create a high biological and chemical oxygen demand and produce conditions conducive to methanogenesis (Quagraine et al., 2005). The largest tailings basin is 10 km2 and effluxes an estimated 100 million l d-1 of methane to the atmosphere, equivalent to about 500 000 cattle (Holowenko et al., 2000). Flux data are not available for all sites, but if the rates are similar, the total flux from all ponds is around 1.7 billion l CH4 d−1. The surface water caps (6–10 m deep) of the ponds, from which recycle water is drawn, are relatively clear and support some aerobic microbial activity. We hypothesized that, similar to a natural wetland, aerobic methanotrophic bacteria are active in the surface water and remove some of the methane produced before it effluxes to the atmosphere.
Known aerobic methanotrophs belong to the Alphaproteobacteria and Gammaproteobacteria classes of the Proteobacteria, the phylum Verrucomicrobia and candidate phylum NC10 (Op den Camp et al., 2009). The proteobacterial methanotrophs have been well-studied in various environments. There are two forms of the key enzyme methane monooxygenase (MMO), a soluble form (sMMO) and a membrane-bound or particulate form (pMMO). The pMMO is universal to all known methanotrophs with the exception of Methylocella spp. and Methyloferula stellata (Dedysh and Dunfield, 2010; Vorob’ev et al., 2011), and the phylogenies of the pmoCAB genes encoding it correspond well to 16S rRNA gene-based phylogeny (Kolb et al., 2003; Op den Camp et al., 2009). Therefore, methanotroph communities are commonly investigated by cultivation-independent recovery and sequence analysis of the pmoA gene (McDonald et al., 2008). Although effective, this technique fails to detect sMMO-only methanotrophs like Methylocella spp., which may be important in some ecosystems, such as northern wetlands (Dedysh and Dunfield, 2010). DNA stable isotope probing (SIP) using 13CH4 has commonly been used in combination with analysis of pmoA or 16S rRNA genes to investigate active methanotrophic communities in various environments (McDonald et al., 2008).
Chemical properties of oilsands tailings water have been summarized elsewhere (Quagraine et al., 2005; Allen, 2008; Penner and Foght, 2010). The water is slightly alkaline (around pH 8.0) due to the use of NaOH in the extraction process, has a high salt content of about 2.2 g l−1 and also has high contents of various metals, sulfides and hydrocarbons. Indigenous methanotrophic species are therefore likely to be different from those found in other subarctic wetlands common in northern Alberta, which are usually acidic (pH 3.5–5), salt-poor oligotrophic habitats (Dedysh, 2011). In the present study, we combined SIP, metagenomics, pyrotag sequencing of 16S rRNA and pmoA genes and pmoA microarray analyses to examine the activity and community structure of aerobic methanotrophic bacteria in the surface layer of oilsands tailings ponds. A tailings-affected wetland and natural bitumen outcrops were studied for comparison.
Methods
Sampling and water chemistry
Surface water (0–10 cm) was sampled at 1–3-month intervals over 2010–2011 from two tailings ponds near Fort McMurray, Alberta, Canada: here designated Pond A and Pond B. These two sites are not isolated, as water is occasionally cycled between them. Samples were taken from a floating barge 10–15 m from shore. Four-liter polycarbonate containers were rinsed three times with water before filling to the top and capping. These were transported to the University of Calgary at ambient temperature and processed for experiments within 7 d of sampling. The chemical properties of the surface waters determined at two dates are summarized in Supplementary Table S1. Water was saline, alkaline and rich in ammonia and sulfides. Dissolved O2 was well below the saturation point of around 9 mg l−1 at the sampling temperatures; the maximum was 0.38 mg l−1 at the air–water surface of Pond A. The pH values were measured for each sample taken over the entire study period and ranged from 7.4–8.7 (mean 8.0).
Other sites were sampled on September 25, 2009 and August 5, 2010 to determine: (i) whether the methanotrophs detected in Ponds A and B were found in tailings waters from another industrial operation, and (ii) whether the same methanotrophs were detected in natural sites. These samples included: surface waters from several tailings ponds operated by a different company; a shallow fen that received tailings input but also fresh runoff water so that it was still vegetated with reeds; and natural bitumen deposits from Saline Creek in Fort McMurray (56°41′N, 111°20′W). As streams erode soils in this region, oilsands deposits are exposed. We removed >10-cm-diameter bitumen blocks from the bed of Saline Creek and scraped off the surface layer (1 mm) to determine aerobic methanotroph communities associated with natural bitumen deposits.
Potential methane oxidation
To concentrate bacteria, 200 ml of fresh tailings water was filtered through a 0.22-μm filter. The filter and 20 ml of tailings water were added to a 100-ml serum bottle (equivalent to 220 ml of tailings water and 80 ml of headspace). Each sample was run in duplicate or triplicate. Vials containing 20 ml of water only were used as abiotic leakage controls. Vials were sealed with butyl rubber stoppers, and CH4 (8–10% v/v) and CO2 (10%) were added to the headspace. Bottles were incubated at 23 °C on a rotary shaker at 180 r.p.m. Headspace methane was monitored at 1-d intervals for 7 d using a Varian 450-GC gas chromatograph equipped with an FID detector (250 °C), and 2 mm × 0.5 m Hayesep N and a 2 mm × 1.2 m Molecular Sieve 16X column in series (70 °C) (Agilent Technologies, Mississauga, ON, Canada). Methane oxidation rates were calculated by linear regression of methane mixing ratios in the vial headspaces.
Pyrotag sequencing of 16S rRNA and pmoA genes
Water samples were filtered as described above, and DNA from the filters extracted using the FastDNA Spin Kit for Soil (MP Biomedicals, Santa Ana, CA, USA), with extra purification steps as described by Knief et al. (2003). 16S rRNA genes were amplified using FLX Titanium amplicon primers 454T_RA_X and 454T_F, comprised of 16S rRNA gene-targeted primers 926f and 1392r at the 3′- ends plus 10-nucleotide (nt) multiplex identifiers and Roche Titanium chemistry adaptors at the 5′- ends (Ramos-Padrón et al., 2011). Barcoded PCR reactions were performed, and the products were purified as described by Sharp et al. (2012). Products were sequenced at the Genome Quebec and McGill University Innovation Centre, Montreal, Quebec with a Genome Sequencer FLX Instrument, using a GS FLX TitaniumSeries Kit XLR70 (Roche Diagnostics Corporation, Laval, QC, Canada). Sixty barcoded samples were multiplexed on a half-chip, yielding on average 10,500 reads per sample. Samples with <3000 reads were deleted. Sequence-read data sets were processed using QIIME (Caporaso et al., 2010) to remove low-quality sequences, dereplicate, cluster operational taxonomic units (OTUs) at 97% identity and classify OTUs via BLAST (Basic Local Alignment Search Tool) against the Greengenes database (DeSantis et al., 2006). The low-quality score threshold was set at 25, the average quality was usually 35. Singleton reads were ignored because of the high probability of chimera formation. Pyrotag data sets have been submitted to the Sequence Read Archive under accession number SRP013946.
pmoA genes for pyrosequencing were amplified as described previously (Knief et al., 2003). The pmoA primers 189f and 682r were modified via the inclusion of Roche Titanium chemistry adaptors and a 10-nt barcode on primer 189f. After pyrotag sequencing analysis of the amplicons, pmoA sequences were quality filtered and clustered at 97% identity using PyroTagger (Kunin and Hugenholtz, 2010), and representative OTU sequences were identified via BLAST.
Representative OTU sequences were aligned to a local copy of the 16S rRNA SILVA database or to an aligned database of public-domain pmoA sequences using ARB (Ludwig et al., 2004). Sequences were manually inspected for chimeric segments and homopolymer errors. For constructing 16S rRNA gene phylogenies, a skeleton tree of nearly complete sequences (>1400 nt) was first constructed and the shorter pyrotag sequences (400–440 nt) subsequently added using the parsimony-add function of ARB. BLAST identifications from QIIME were corrected where necessary based on this phylogenetic procedure.
pmoA microarray analysis
pmoA microarray analysis was performed on two samples of Pond B from October and November of 2010 and one sample of Pond A from October, 2010. Amplification of pmoA genes was performed via a two-step PCR. The first-stage PCR amplification used primer set 189f/661r or 189f/682r (McDonald et al., 2008). Thermocycling conditions were: 95 °C for 5 min; followed by 15 cycles of 95 °C for 1 min, 56 °C for 1 min and 72 °C for 1 min; and a final extension at 72 °C for 10 min. Each PCR reaction mixture (50 μl) contained 1 × FailSafe PCR PreMix F (Epicentre Biotechnologies, Madison, WI, USA), 1.5 pmol of each primer and 1 U of Taq DNA Polymerase (Fermentas, Pittsburgh, PA, USA). To each PCR reaction from the first stage, 15 pmol of each primer 189f and T7-661r or T7-682r and an additional 1 U of Taq DNA Polymerase (Fermentas) was added for a second-stage PCR amplification of 25 cycles at 58 °C annealing temperature. PCR products were purified with a QIAquick PCR Purification Kit (Qiagen, Toronto, ON, Canada) and analyzed on a pmoA microarray as described by Stralis-Pavese et al. (2011).
SIP and SIP metagenomics
Seven separate SIP incubations were made, using tailings water sampled from Ponds A and B on August 15, 2010. Tailings water was processed as in the methane oxidation rate experiments, except that bottles were injected with 10% v/v 12CO2 and 1% 13CH4 (99 atom%, Sigma-Aldrich, Oakville, ON, Canada). After 6–10 d cells were collected by centrifugation for 5 min at 25 900 × g. DNA was extracted and stored at −20 °C. DNA was separated by isopycnic centrifugation in CsCl and fractionated exactly as described by Sharp et al. (2012). Selected heavy and light DNA fractions were used for pyrotag analysis as described above. DNA extracted directly from the same tailings water samples was used as a control to determine the expected position of unlabeled DNA in CsCl gradients.
Five microliters of a selected ‘heavy’ SIP fraction was amplified via whole-genome amplification using the REPLI-g UltraFast Mini Kit (Qiagen), yielding 4.7 μg DNA. The Phi29 DNA polymerase was thermally inactivated, and the product sequenced without additional purification. Paired-end DNA libraries were sequenced for 150 cycles using an Illumina HiSeq2000 (Illumina, Inc., San Diego, CA, USA) at Genome Quebec. Raw Illumina reads were passed through an in-house quality control program to filter out known Illumina artifacts, specifically reads that: (i) contained spike-in PhiX sequence; (ii) were shorter than 50 bp after clipping off the partial primer, adapters and the low-quality range at the ends; or (iii) were of low complexity. Duplicated read pairs were identified and consolidated into a single consensus read pair (Hess et al., 2011). Reads that passed the quality control stage were assembled using SOAPdenovo (V1.05, BGI, Shenzhen, China). As no optimal k-mer length exists for any de novo metagenome assembly, the multiple k-mer method was used to obtain the final assembly (Surget-Groba and Montoya-Burgos, 2010). We performed eight assemblies using different values of k-mer size (59, 63, 67, 71, 75, 79, 83 and 87). The different assembles were merged pair-wise using Newbler (V2.6, Roche) and AMOS minimus2 (Sommer et al., 2007) to get the final assembly.
A database of genes involved in methane and methanol oxidation, including formaldehyde oxidation (tetrahydrofolate and tetrahydromethanopterin pathways), formaldehyde fixation (Serine cycle, RuMP cycle) and alleviation of stress caused by ammonia cooxidation by MMO was assembled from both Alphaproteobacteria and Gammaproteobacteria methanotrophs (Ward et al., 2004; Chistoserdova et al., 2009; Vuilleumier et al., 2009; Stein and Klotz, 2011). The metagenome assembly was searched for these genes via local BLAST. Where possible, the BLAST query was taken from the genome of Methylococcus capsulatus, as this is the closest relative with a fully sequenced genome to the predominant methanotroph detected in the surface tailings water.
Results
Potential methane oxidation
Methane oxidation in surface tailings water from Ponds A and B began without a lag phase, indicating that the methanotrophs were active when sampled (Supplementary Figure S1). The initial methane oxidation rates were linear and ranged from 75–152 nmol CH4 ml−1 d−1 (Table 1). Rates were quite consistent over the different sampling times.
Community analyses via 16S rRNA gene pyrotag sequencing
Bacterial communities identified based on 16S rRNA gene pyrotag sequencing are summarized in Supplementary Table S2. The surface of the bitumen samples was populated by OTUs similar to known aerobes, particularly gammaproteobacterial methanotrophs. By contrast, surface waters of the tailings ponds included presumed obligate aerobes (for example, members of the genera Methylocaldum, Xanthobacter, Flavobacterium), obligate anaerobes (for example, of the genera Methanoregula, Methanosarcina, or the order Clostridiales) and facultative anaerobes (for example, in the family Comamonadaceae) as predominant groups. The majority of bacteria detected were Proteobacteria, although one predominant OTU in Pond A was a member of the phylum Acidobacteria. Although this acidobacterium was identified by the Greengenes BLAST as related to the phototroph genus Chloracidobacterium, it is more similar (93%) to some organoheterotrophic strains like Ellin6075 (Joseph et al., 2003). Most predominant OTUs in the tailings ponds surface water belonged to the Betaproteobacteria, particularly to the families Comamonadaceae and Rhodocyclaceae. Most of the OTUs identified to the genus level belonged to genera known to contain hydrocarbon degraders, for example, Acinetobacter, Geobacter, Rhodoferax, Methylibium, Hydrogenophaga, Pseudomonas, Xanthobacter and Acidovorax, as well as some methanogens and methanotrophs (Prince et al., 2010).
Methanotrophs identified via 16S rRNA gene pyrotag sequencing
Most known methanotrophs fall into deeply rooted monophyletic clades with no closely related non-methanotrophic neighbors, and therefore 16S rRNA gene sequence phylogeny can reliably predict methanotrophic function. An aerobic methanotroph belonging to the Methylococcus/Methylocaldum cluster of Gammaproteobacteria (OTU 12 103) was among the predominantly detected OTUs in Pond A, making up on average 1.5% of all reads (Figure 1; Supplementary Table S2C). This OTU was in fact the predominant methanotroph detected in all surface tailings waters and all sampling times (Table 1). A few other methanotrophs were detected at lower relative abundance (Table 1; Figure 1), including members of the genera Methylomonas, Crenothrix/Methylosoma and Methylobacter. The genera Methylococcus and Methylomicrobium were detected only in Pond B. The methanotrophic communities in the bitumen and fen samples were distinct from those in the surface tailings waters, indicating that the unique chemistry of the tailings water selected for a particular community. Methylocaldum strains were not detected in the bitumen samples from Saline Creek. Instead the major methanotrophs belonged to the Crenothrix/Methylosoma cluster (Supplementary Table S2A, Figure 1). Some of the OTU’s in this group were found only in the bitumen samples. In the tailings-affected fen, only Methylobacter spp. were detected. Few alphaproteobacterial methanotrophs were detected in any sample. Of 252 917 total reads, there were 2 reads of a Methylocystis sp. and 13 reads of potential methanotrophs in the family Beijerinckiaceae.

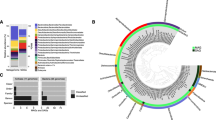
Phylogenetic tree based on 16S rRNA gene sequences of methanotrophic Proteobacteria. To create the most reliable phylogeny, a skeleton tree was constructed from nearly complete reference sequences (>1400 bp) via Neighbor-joining with a Jukes-Cantor correction and 10 000 bootstraps. Shorter pyrotag sequences and sequences from the SIP-metagenome assembly (in bold) were then added to the skeleton tree using the parsimony-add function of ARB. For comparison, the total number of reads of each OTU in all samples is given in parentheses (entire data set was 252 917 reads), and the key Methylocaldum sp. OTU is in a box. Some major groups of methanotrophs are indicated at the right. The scale bar represents 0.1 change per nucleotide position. Bootstrap values >70% are indicated for key nodes.
The predominance of methanotrophs in the pyrotag libraries was not correlated to the methane oxidation rates (Table 1), suggesting that these read ratios reflect primarily population levels of other bacteria: that is, fluctuating populations of other bacteria will cause the ratio of methanotroph reads to change even if the methanotroph population is constant. The difference could also reflect differing ratios of active to inactive bacteria within the methanotroph community.
Although most groups identified in Table 1 fall within known clusters of obligate methanotrophs, two groups were difficult to assign. The first comprised sequences affiliated to the Beijerinckiaceae family, which contains obligate methanotrophs like Methylocapsa acidiphila, facultative methanotrophs like Methylocella silvestris and non-methanotrophs like Beijerinckia indica (Dedysh and Dunfield, 2010). All are closely related (<4% 16S rRNA divergence). The role of such OTUs in methane oxidation is therefore unknown, although they made up a small proportion of the methanotroph reads and are probably of minor importance. The second problematic OTU was identified by QIIME as belonging to the family Methylococcaceae but was equally similar to the Thiococcus/Thiocapsa group of Chromatiaceae. Several different phylogenetic constructions failed to strongly support the affiliation of this cluster with either group (data not shown). This OTU was nearly as abundant as the Methylocaldum OTU (on average 72% as many reads). However, it did not enrich in SIP experiments and no unknown pmoA sequences were found that would be expected to belong to this group. The group probably does not consist of methanotrophs.
Many putatively methylotrophic species were present in the surface tailings water, such as Betaproteobacteria of the genus Methyloversatilis and family Methylophilaceae, as well as Alphaproteobacteria of the genera Xanthobacter, Hyphomicrobium and Methylobacterium (Supplementary Tables S2B and C). These could feed partially on byproducts of methane oxidation, particularly methanol or formaldehyde.
Methanotrophic community analyses via pmoA pyrotag sequencing and microarray analyses
Two samples of Pond B and one of Pond A were used for additional analyses of methanotroph communities via pmoA pyrotag sequencing and pmoA microarray analysis. Table 2 summarizes the methanotrophs detected by all the three methods. Many genera of Gammaproteobacteria methanotrophs (Methylobacter/Methylomicrobium/Crenothrix/Methylosoma/ Clonothrix/Methylosarcina/Methylovulum—together sometimes called the type 1A group) are closely related, and it is difficult to map pmoA to 16S rRNA gene sequences among these genera. pmoA sequences that fell into this group were therefore combined. A single pmoA sequence comprised up to 98% of all pmoA pyrotag sequencing reads. This sequence was related to the Methylococcus/Methylocaldum cluster phylogenetically (Figure 2) but was <90% identical to pmoA genes of cultured species. Given its predominance, this pmoA sequence likely belongs to OTU 12 103 identified as a Methylocaldum sp. via 16S rRNA sequencing (Figure 1). Microarray analysis was performed with two pmoA primer sets 189f/682r and 189f/661r (Supplementary Figure S2). The analysis verified the predominance of Methylocaldum (probes Mcl 404, Mcl 408) and/or Group 501 (a cluster related to Methylocaldum/Methylococcus, probes 501–375, 501–286). This positive detection may represent cross-hybridization of these probes with our Methylocaldum-like bacterium, as this sequence showed few mismatches to these probes (2 and 1, respectively). The genera Methylomonas (Mm275) and Methylobacter (Mb380, Mb271, Mb_SL#3-300) were detected strongly in all samples with the 189f/661r primer set but were less represented when using the 189f/682r primer pair. Strong signals for the genus Methylosinus (Msi423, Msi294, Msi232) and weak signals for Methylocystis (Mcy522, McyM309) were detected in Pond B only.

Phylogenetic tree based on partial-derived PmoA sequences of Proteobacteria (165 amino acids). The tree was constructed via Neighbor-joining with 10 000 bootstraps. Sequences detected in surface tailings water via PCR analysis or in the metagenomic assembly from the heavy DNA fraction of a SIP experiment are indicated in bold. Major known clusters of PmoA and AmoA are indicated at the right. The scale bar represents 0.1 change per position. Bootstrap values >80% are indicated. The predominant Methylocaldum sp. OTU is in a box. The PxmA group represents a second copy of a PmoA-like sequence of unknown function recently found in the genomes of some Gammaproteobacteria methanotrophs. The tree was rooted against a set of eight AmoA sequences from Betaproteobacteria.
The three analyses agreed well, identifying the most abundant species as a Methylocaldum sp., followed by Methylomonas spp., and other Gammaproteobacteria. The largest discrepancy was in the pmoA pyrotag sequencing results, which appeared to under-represent some Gammaproteobacteria and over-represent Alphaproteobacteria compared with the other two methods. This was probably due to the 682r primer used for pmoA pyrosequencing, which is known to bias against Gammaproteobacteria (McDonald et al., 2008).
SIP
Five SIP incubations with Ponds A and B sampled on August 15, 2010 showed similar results. These are summarized in Supplementary Table S3, and an example SIP experiment is shown in Figure 3 (1% CH4, Pond B, 10 d). Compared with DNA extracted directly from tailings water, the 13CH4-incubated sample showed a long tail of heavy DNA (Figure 3). Based on 16S rRNA pyrotag sequencing, the proportion of methanotrophs was much higher in the heavy DNA compared with the light DNA or to the raw tailings water DNA (Figure 3). Methylocaldum spp. and Methylomonas spp. were the predominant 13C-labeled OTUs, followed by Methylobacter and Methylomicrobium (Supplementary Table S3). A large amount of Methylophilales was also labeled, probably as a result of cross-feeding via methanol. In two additional SIP incubations an amoeba (98% sequence identity to Protacanthamoeba bohemica) was strongly detected in heavy DNA (up to 40% of all reads), indicating that it was grazing on biomass ultimately derived from methanotrophy, most likely directly on methanotrophic bacteria. Grazing by amoebae has previously been observed in soils with elevated methanotroph populations (Murase and Frenzel, 2008). These two heavily grazed SIP experiments were not included in Supplementary Table S3, as the methanotroph species ratios may have been affected by selective grazing.

DNA density gradients and methanotroph communities based on 16S rRNA pyrotag sequencing for a SIP incubation using Pond B surface tailings water. Incubation was under 1% v/v 13CH4 for 10 d. The density profile and methanotrophic community of fresh tailings water is given for comparison. The density profiles were normalized by setting the fraction with the highest concentration to 1. For the SIP, the community from ‘heavy’ DNA of 1.75 g ml−1 is compared with that of ‘light’ DNA from 1.72 g ml−1. Only methanotrophic groups and abundant methylotrophs in the family Methylophilaceae are shown in the pie charts. White pie sections represent all other bacteria.
SIP metagenomics
A metagenome analysis was performed on the heavy SIP fraction indicated in Figure 3. Illumina sequencing yielded 382 million read pairs amounting to 38.2 Gbp. De novo assembly of the quality-controlled metagenomic reads produced 34 Mb in contigs >200 bp, with an N50 of 632 bp. Only 2.6 Mb were on contigs >5 kB length. Because the assembly was poor, analysis was limited to detecting particular genes. Conclusions about the species possessing these genes and possible gene linkages were not possible.
The metagenome verified the main methanotroph species present via a non-PCR method. 16S rRNA genes detected in the metagenome assembly included members of the key genera Methylocaldum and Methylomonas, as well as Methylomicrobium and Methylosoma/Crenothrix (Figure 1). A nearly complete pmoCAB operon was assembled that was related to a Methylocaldum sp. (Supplementary Table S4; Figure 2). This operon showed the same gene arrangement as most methanotrophs (pmoCAB) and an overall identity of 88% to the operon of Methylococcus capsulatus and 87% to the operon of Methylocaldum szegediense. In addition, partial pmoA gene sequences from a Methylomonas sp. and a Methylomicrobium sp. were found. Two deeply branching pmoA-like genes detected in the metagenome were not detected via PCR (Figure 2).
Although pmoCAB and 16S rRNA genes were found in the metagenome for the key methanotrophs, no genes (mmoXYBZD) encoding sMMO were found using a BLAST threshold of e−5. Diverse genes encoding formaldehyde utilization pathways were detected, including hps encoding the key enzyme hexulose phosphate synthase of the Ribulose monophosphate pathway for formaldehyde assimilation, and mtdB encoding methylene-H4MPT dehydrogenase for formaldehyde oxidation. Nitrogen metabolism genes common to methanotrophs were detected, including haoA encoding hydroxylamine oxidoreductase, and norB encoding nitric oxide reductase (Stein and Klotz, 2011). Most matched closely to Gammaproteobacteria methanotrophs, although some of the norB assemblies matched closely to genes from methylotrophic Betaproteobacteria. Overall, most methylotrophy genes, when analyzed via BLAST against the non-redundant protein database, matched most closely to genome sequences of Methylomonas methanica and Methylococcus capsulatus, or less frequently to Methylobacter spp. and Methylomicrobium spp. The same Gammaproteobacteria methanotrophs were therefore identified in the shotgun metagenome data that were identified via PCR-based pmoA and 16S rRNA gene analyses. Additionally, some methylotrophy genes were probably derived from betaproteobacterial methylotrophs growing via cross-feeding of methanol. There is thus a good agreement of the metagenome with the PCR results.
Discussion
The potential methane oxidation rates in surface tailings waters of the two ponds were as high as 152 nmol CH4 ml−1 d−1. This is within the range of rates seen in natural lake and ocean waters (for example, Bastviken et al., 2002; Carini et al., 2005; Krüger et al., 2005; He et al., 2012), although soils and sediments in similar climates can show much higher rates, ranging from 0.17–80 μmol CH4 g−1 d−1 (Kravchenko, 2002; Wagner et al., 2005; Kip et al., 2010; Graef et al., 2011; Lee et al., 2011; Barbier et al., 2012; He et al., 2012). The aeration depth of the ponds was estimated as 60 cm for Pond B and 1.5 m for Pond A (Supplementary Table S1). Methanotrophs have a high affinity for O2, with Ks values of 2–10 μM, and can even outcompete heterotrophs for O2 at very low concentrations (van Bodegom et al., 2001), so they are probably active where any detectable O2 is present. Based on a conservative estimate of a 50-cm active layer, 150 nmol CH4 ml−1 d−1 extrapolates over the entire Pond B (10 km2) to about 17 × 106 l CH4 d−1, which is 17% of the estimated CH4 emission rate in this site (Holowenko et al., 2000). This extrapolation is obviously crude and intended as a framework figure only. The key uncertainty is the depth of aeration, which is likely variable in time and space. The oxygen demand will vary with the input of fresh tailings and the removal of recycled surface water, as well as with natural factors like temperature. More intense monitoring of the O2 budget would be valuable. Much of the methane flux is via ebullition (Holowenko et al., 2000), and this methane will be largely unavailable to methanotrophs. However, the data demonstrate that methanotrophs have the potential to reduce methane efflux from these sites.
The predominance of methanotrophs within the microbial community as a whole varied from <0.01% to 5.8% of total reads. However, the species composition of the methanotrophic community changed very little, and a single OTU predominated in all surface tailings water samples. This was a gammaproteobacterium related to the genus Methylocaldum, although based on its 16S rRNA sequence distance to type strains (7.6–8.7% based on the metagenome sequence spanning Escherichia coli positions 758–1527) it should represent a new genus. A variety of detection techniques verified its predominance. Although these techniques were not strictly quantitative, they were based on two genes, multiple primer sets, and both pyrosequencing and hybridization detection methods. It is exceedingly unlikely that all of these techniques would show the same bias to a single organism. Activity-based SIP verified the importance of the Methylocaldum sp. Other species are capable of living in these waters, and members of the genera Methylomonas, Methylobacter and Methylomicrobium grew rapidly in SIP experiments using surface tailings water. However, these species seemed to be less abundant in situ (Table 1, Figure 3). The constancy of the methanotrophic community is remarkable given that the sample set included ponds from different industrial operations and two particular ponds sampled periodically over 2 years.
This predominance of the Methylocaldum sp. was limited to the surface tailings water. Communities associated with a fen and natural bitumen outcrops in the same geographical region were dominated by strains related to the genera Methylobacter and Crenothrix/Methylosoma, respectively. We did not detect a pmoA sequence branching distantly to other proteobacterial pmoAs that is proposed to belong to Crenothrix polyspora (Stoecker et al., 2006), so the latter OTU probably represents a phylotype similar to a Methylosoma sp. Non-petroleum-influenced ecosystems in the area were not analyzed in this study, but there is already a large body of work available on northern peatlands like those found in the Fort McMurray area. Enumeration via species-specific 16S rRNA-targeted fluorescent oligonucleotide probes and cultivation-independent recovery of pmoA and mmoX genes have together demonstrated that the most important species in peatland ecosystems are strains of Alphaproteobacteria in the genera Methylocystis, Methylocella and Methylocapsa (Dedysh et al., 2001, 2003; Dunfield, 2009), as well as some Gammaproteobacteria related to Methylomonas (Kip et al., 2011, 2012). The Alphaproteobacteria have been isolated and shown to be mild acidophiles and, in the case of the genera Methylocella and Methylocapsa, adapted to oligotrophic salt-poor waters (Dunfield, 2009; Dunfield and Dedysh, 2010). These conditions are obviously dramatically different from the alkaline, slightly saline tailings water, where Alphaproteobacteria methanotrophs were almost undetectable. Alkaline and saline waters tend to be dominated by Gammaproteobacteria methanotrophs, particularly strains of Methylomicrobium (Trotsenko and Khmelenina, 2002; Lin et al., 2004; Dunfield, 2009). Methane SIP of Lonar lake (pH 10; 0.9% salt) identified a Methylomicrobium sp. as the predominant methanotroph (Antony et al., 2010). Gammaproteobacteria related to the genera Methylobacter and Methylococcus/Methylocaldum and some Alphaproteobacteria related to Methylocystis were detected via SIP in pH 9.4 soil from a coal mine (Han et al., 2009). A predominance of gammaproteobacterial methanotrophs in tailings water is reasonable based on these studies, although it is rare to find a community dominated by Methylocaldum spp.
The predominance of the Methylocaldum and other Gammaproteobacteria methanotrophs may therefore be influenced by the high pH and salinity of the tailings water. Other factors known to influence methanotrophic communities are temperature and the availability of N, Cu and O2 (Dunfield, 2009; Dedysh and Dunfield, 2010; Semrau et al., 2010). Some strains of the genera Methylocaldum and Methylococcus grow optimally at 40–60°C (Dunfield, 2009), so a potential explanation for their predominance is that they survive in high-temperature process water. The surface water of the tailings ponds is constantly removed, heated to 60 °C and reused in the bitumen extraction process before being returned into the pond systems. Aerobic organisms in the surface water layer must either grow rapidly enough to maintain their populations against this constant water removal, much like growth in a chemostat, or they must survive the heating process. Nitrogen is not limited in the ponds, in fact some nitrifying bacteria were detected (Supplementary Figure S2). However, the high ammonia/ammonium concentrations up to 17 mg l−1 should require that methanotrophic species have mechanisms to deal with the toxic byproducts of ammonia oxidation by pMMO (Stein and Klotz, 2011). Genes encoding for hydroxylamine reductase and nitric oxide reductase were detected in the metagenome. Most were closely related to genes found in strains of the genera Methylomonas, Methylotenera and Methylomicrobium. This does not necessarily mean that these genes do not derive from the Methylocaldum OTU, but this species may be using other mechanisms for handling nitrogen stress. O2 is clearly a limiting factor in the surface tailings water, and different O2 concentrations have been shown to control methanotroph community compositions, although there are no consistent trends in what species predominate under high CH4 and low O2 conditions like those in the tailings water. They can be different Alphaproteobacteria or Gammaproteobacteria species (Amaral and Knowles, 1995; Henckel et al., 1999; Henckel et al., 2000, van Bodegom et al., 2001; Horz et al., 2002; Pester et al., 2004; Bussmann et al., 2006). Methane and O2 levels certainly affect methanotroph communities, but the effect is probably site-specific and difficult to generalize.
Probably because of whole-genome amplification biases, the SIP-metagenome assembly was poor, but it did supplement the PCR-based analysis of the SIP experiment. A group of pmoA-like sequences recently detected as part of a second pmo operon in some methanotrophic Gammaproteobacteria (Tavormina et al., 2011) was found only in the metagenome (PxmA group in Figure 2). PCR biases make these undetectable with standard pmoA primer sets (Tavormina et al., 2008), but the use of metagenomics allowed us to find them in the tailings water. This constitutes the first detection of these genes outside a marine system (Tavormina et al., 2008). The function of these pmoA-like genes is presently unknown.
The community also contained other genes typical of gammaproteobacterial methanotrophs (Supplementary Table S4). However, genes encoding sMMO were not found. Many methanotrophs encode this enzyme in addition to pMMO. Although the Illumina run would theoretically allow 100–1000 × coverage of a simple community containing <10 genomes, biases connected to the method and to the whole-genome amplification step mean that some genes may have been missed. Nevertheless, pmo and 16S rRNA genes were detected for the major species in the metagenome and therefore the most likely explanation for the failure to detect any of the mmo operon genes is that they were simply not present. Described Methylocaldum species do not encode sMMO, but some Methylococcus spp. and Methylomonas spp. do, so the result is surprising. The lack of sMMO in the surface tailings water community might have biotechnological implications for co-oxidation of pollutants by methanotrophs, as sMMO has a broader substrate range than pMMO (Dalton, 2005; Semrau, 2011). One reason for the absence of methanotrophs possessing sMMO may be that this enzyme would rapidly generate toxic byproducts from the oxidation of the diverse aromatic hydrocarbons in the water and thereby damage the methanotrophs. Semrau (2011) noted that, while both pMMO-expressing and sMMO-expressing methanotrophs degrade diverse pollutants, the sMMO-expressing cells do so at a higher rate and hence do not survive as well in complex contaminant mixtures. Another explanation for the predominance of pMMO might be the large concentrations of Cu in the tailings water, reported as up to 0.005 mg l−1 (Quagraine et al., 2005; Kelly et al., 2010). sMMO is generally only expressed in low-Cu conditions (Semrau et al., 2010), so an ecosystem rich in Cu might render sMMO as a backup to pMMO unnecessary and select for pMMO-only methanotrophs.
Surface tailings water communities detected via 16S rRNA gene pyrotag sequencing were an unusual mixture of obligate aerobes, obligate anaerobes and facultative anaerobes. The chemical and biological oxygen demands in the ponds are very high due to the hydrocarbons and sulfides present, so obligate aerobes like methanotrophs are only active in the surface waters. Deeper anaerobic layers of the ponds are dominated by methanogenic Euryarchaeota and other obligate anaerobes such as Clostridia (Penner and Foght, 2010; Ramos Padron et al., 2011). These were also detected in surface waters, as were several OTUs that should represent obligate aerobes. The abundant members of the genus Flavobacterium, phylum Acidobacteria and family Rhodospirillaceae in the 16S rRNA gene pyrotag read sets (Supplementary Tables 2B–D) were also abundant in the light fractions of SIP experiments (usually >20% of all reads, data not shown) after aerobic shaking for 6–10 d, which verified that these OTUs represented fully aerotolerant bacteria. However, by far the predominant OTUs in surface tailings water were Betaproteobacteria, particularly in the families Comamonadaceae and Rhodocyclaceae, which are likely to be facultative anaerobes. These families are known to be diverse metabolically, containing species capable of phototrophy, lithotrophy (S and H2), organotrophy, aerobic respiration, fermentation, Fe3+ reduction and denitrification (Garrity et al., 2001; Willems and Gillis, 2001). Many degrade hydrocarbon pollutants (Prince et al., 2010). The surface water is very low in O2 and is subject to constant forced circulation through the addition of fresh tailings and drawoff of recycle water, allowing mixing of aerobic and anaerobic communities. This would explain the predominance of facultative anaerobes like the Comamonadaceae, as a versatile, primarily anaerobic metabolism would be favored in such a system.
Our results provide a first glimpse of the methanotrophic potential of surface waters of oilsands tailings ponds. Potential rates of methanotrophy are high and the methanotroph communities are predominated by a few Gammaproteobacteria that appear to have pMMO but no sMMO, particularly a Methylocaldum-like species. Environmental factors selecting for these species may be the alkaline pH, high salinity, high ammonia, high Cu, low O2, high concentrations of co-oxidisable substrates and occasionally high temperatures. Study of the predominant species after isolation should elucidate their adaptations. Given the accelerating development of the oilsands resource, and concurrent global concerns about its carbon footprint (Swart and Weaver, 2012), it is important to understand the carbon cycle of this system (Rooney et al., 2012). The most obvious limiting factor to methanotrophy in situ is O2 limitation. Because of the large chemical and biological O2 demands of the water and the large pollutant loads, these lakes are fundamentally different from natural systems. Engineering a deeper, less turbid surface water cap might allow better O2 penetration and foster the growth of the aerobic community, including methanotrophs. This would be desirable to increase degradation rates of harmful chemicals and reduce the large methane emissions associated with tailings ponds.
Accession codes
References
Allen EW . (2008). Process water treatment in Canada’s oil sands industry: II. A review of emerging technologies. J Environ Eng Sci 7: 499–524.
Amaral JA, Knowles R . (1995). Growth of methanotrophs in methane and oxygen counter gradients. FEMS Microbiol Lett 126: 215–220.
Antony CP, Kumaresan D, Ferrando L, Boden R, Moussard H, Fernandez-Scavino A et al. (2010). Active methylotrophs in the sediments of Lonar Lake, a saline and alkaline ecosystem formed by meteor impact. ISME J 4: 1470–1480.
Barbier BA, Dziduch I, Liebner S, Ganzert L, Lantuit H, Pollard W et al. (2012). Methane-cycling communities in a permafrost-affected soil on Herschel Island, Western Canadian Arctic: active layer profiling of mcrA and pmoA genes. FEMS Microbiol Ecol 82: 287–302.
Bastviken D, Ejlertsson J, Tranvik L . (2002). Measurement of methane oxidation in lakes: a comparison of methods. Environ Sci Technol 36: 3354–3361.
Bussmann I, Rahalkar M, Schink B . (2006). Cultivation of methanotrophic bacteria in opposing gradients of methane and oxygen. FEMS Microbiol Ecol 56: 331–344.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Carini S, Bano N, LeCleir G, Joye SB . (2005). Aerobic methane oxidation and methanotroph community composition during seasonal stratification in Mono Lake, California (USA). Environ Microbiol 7: 1127–1138.
Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME . (2009). The expanding world of methylotrophic metabolism. Annu Rev Microbiol 63: 477–499.
Dalton H . (2005). The Leeuwenhoek lecture 2000. The natural and unnatural history of methane-oxidizing bacteria. Phil Trans Royal Soc B: Biol Sci 360: 1207–1222.
Dedysh SN, Derakshani M, Liesack W . (2001). Detection and enumeration of methanotrophs in acidic Sphagnum peat by 16S rRNA fluorescence in situ hybridization, including the use of newly developed oligonucleotide probes for Methylocella palustris. Appl Environ Microbiol 67: 4850–4857.
Dedysh SN, Dunfield PF, Derakshani M, Stubner S, Heyer J, Liesack W . (2003). Differential detection of type II methanotrophic bacteria in acidic peatlands using newly developed 16S rRNA-targeted fluorescent oligonucleotide probes. FEMS Microbiol Ecol 43: 299–308.
Dedysh SN, Dunfield PF . (2010). Facultative methanotrophs. In: Timmis KN (eds) Handbook of Hydrocarbon and Lipid Microbiology. Springer: Berlin, pp 1967–1976.
Dedysh SN . (2011). Cultivating uncultured bacteria from northern wetlands: knowledge gained and remaining gaps. Front Microbiol 2: 184.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Dunfield PF . (2009). Methanotrophy in extreme environments. In: Battista J (eds) Encyclopedia of Life Sciences. John Wiley and Sons Ltd: Chichester.
Dunfield PF, Dedysh SN . (2010). Acidic environments. In: Timmis KN (ed.) Handbook of Hydrocarbon and Lipid Microbiology. Springer: Berlin, pp 2181–2192.
Energy Resources Conservation Board (2012) ST98-2011: Alberta’s Energy Reserves 2011 and Supply/Demand Outlook 2012-2021. http://www.ercb.ca/RSS/yp-starter/401.aspx.
Garrity GM, Bell JA, Lilburn T . (2001). Family I. Rhodocyclaceae fam. nov. In: Bergey DH, Boone DR, Garrity GM, Castenholz RW, Brenner DJ, Krieg NR, Staley JT (eds) Bergey’s Manual of Systematic Bacteriology: The Proteobacteria: Part C. The Alpha-, Beta-, Delta-, and Epsilonproteobacteria. Springer: Berlin, pp 887.
Government of Alberta (2012) Oil sands tailings factsheet http://www.oilsands.alberta.ca/FactSheets/Tailings_FSht_June_2012_Online.pdf.
Graef C, Hestnes AG, Svenning MM, Frenzel P . (2011). The active methanotrophic community in a wetland from the High Arctic. Environ Microbiol Rep 3: 466–472.
Han B, Chen Y, Abell G, Jiang H, Bodrossy L, Zhao J et al. (2009). Diversity and activity of methanotrophs in alkaline soil from a Chinese coal mine. FEMS Microbiol Ecol 70: 196–207.
He R, Wooller MJ, Pohlman JW, Quensen J, Tiedje JM, Leigh MB . (2012). Diversity of active aerobic methanotrophs along depth profiles of arctic and subarctic lake water column and sediments. ISME J 6: 1937–1948.
Henckel T, Friedrich M, Conrad R . (1999). Molecular analyses of the methane-oxidizing microbial community in rice field soil by targeting the genes of the 16S rRNA, particulate methane monooxygenase, and methanol dehydrogenase. Appl Environ Microbiol 65: 1980–1990.
Henckel T, Roslev P, Conrad R . (2000). Effects of O2 and CH4 on presence and activity of the indigenous methanotrophic community in rice field soil. Environ Microbiol 2: 666–679.
Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331: 463–467.
Holowenko FM, MacKinnon MD, Fedorak PM . (2000). Methanogens and sulfate-reducing bacteria in oil sands fine tailings waste. Can J Microbiol 46: 927–937.
Horz HP, Raghubanshi AS, Heyer J, Kammann C, Conrad R, Dunfield PF . (2002). Activity and community structure of methane-oxidising bacteria in a wet meadow soil. FEMS Microbiol Ecol 41: 247–257.
Joseph SJ, Hugenholtz P, Sangwan P, Osborne CA, Janssen PH . (2003). Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl Environ Microbiol 69: 7210–7215.
Kasperski KL, Mikula RJ . (2011). Waste streams of mined oil sands: characteristics and remediation. Elements 7: 387–392.
Kelly EN, Short JW, Schindler DW, Hodson PV, Ma M, Kwan AK et al. (2009). Oil sands development contributes polycyclic aromatic compounds to the Athabasca river and its tributaries. Proc Natl Acad Sci USA 106: 22346–22351.
Kelly EN, Schindler DW, Hodson PV, Short JW, Radmanovich R, Nielsen CC . (2010). Oil sands development contributes elements toxic at low concentrations to the Athabasca river and its tributaries. Proc Natl Acad Sci USA 107: 16178–16183.
Kip N, Dutilh BE, Pan Y, Bodrossy L, Neveling K, Kwint MP et al. (2011). Ultra-deep pyrosequencing of pmoA amplicons confirms the prevalence of Methylomonas and Methylocystis in Sphagnum mosses from a dutch peat bog. Environ Microbiol Rep 3: 667–673.
Kip N, Fritz C, Langelaan ES, Pan Y, Bodrossy L, Pancotto V et al. (2012). Methanotrophic activity and diversity in different Sphagnum magellanicum dominated habitats in the southernmost peat bogs of Patagonia. Biogeosciences 9: 47–55.
Kip N, Van Winden JF, Pan Y, Bodrossy L, Reichart GJ, Smolders AJP et al. (2010). Global prevalence of methane oxidation by symbiotic bacteria in peat-moss ecosystems. Nature Geosci 3: 617–621.
Knief C, Lipski A, Dunfield PF . (2003). Diversity and activity of methanotrophic bacteria in different upland soils. Appl Environ Microbiol 69: 6703–6714.
Kolb S, Knief C, Stubner S, Conrad R . (2003). Quantitative detection of methanotrophs in soil by novel pmoA-targeted real-time PCR assays. Appl Environ Microbiol 69: 2423–2429.
Kravchenko IK . (2002). Methane oxidation in boreal peat soils treated with various nitrogen compounds. Plant Soil 242: 157–162.
Krüger M, Treude T, Wolters H, Nauhaus K, Boetius A . (2005). Microbial methane turnover in different marine habitats. Paleogeog Palaeoclimatol Palaeoecol 227: 6–17.
Kunin V, Hugenholtz P . (2010). PyroTagger: a fast, accurate pipeline for analysis of rRNA amplicon pyrosequence data. The Open J 1: 1–8.
Lee S-Y, Lee SH, Jang JK, Cho K-S . (2011). Comparison of methanotrophic community and methane oxidation between rhizospheric and non-rhizospheric soils. Geomicrobiol J 28: 676–685.
Lin JL, Radajewski S, Eshinimaev BT, Trotsenko YA, McDonald IR, Murrell JC . (2004). Molecular diversity of methanotrophs in Transbaikal soda lake sediments and identification of potentially active populations by stable isotope probing. Environ Microbiol 6: 1049–1060.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
McDonald IR, Bodrossy L, Chen Y, Murrell JC . (2008). Molecular ecology techniques for the study of aerobic methanotrophs. Appl Environ Microbiol 74: 1305–1315.
Murase J, Frenzel P . (2008). Selective grazing of methanotrophs by protozoa in a rice field soil. FEMS Microbiol Ecol 65: 408–414.
Op den Camp HJM, Islam T, Stott MB, Harhangi HR, Hynes A, Schouten S et al. (2009). Minireview: environmental, genomic, and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ Microbiol Rep 1: 293–306.
Penner TJ, Foght JM . (2010). Mature fine tailings from oil sands processing harbour diverse methanogenic communities. Can J Microbiol 56: 459–470.
Pester M, Friedrich MW, Schink B, Brune A . (2004). pmoA-based analysis of methanotrophs in a littoral lake sediment reveals a diverse and stable community in a dynamic environment. Appl Environ Microbiol 70: 3138–3142.
Prince RC, Gramain A, McGenity TJ . (2010). Prokaryotic hydrocarbon degraders. In: Timmis KN (eds) Handbook of Hydrocarbon and Lipid Microbiology. Springer: Berlin, pp 1669–1692.
Quagraine EK, Peterson HG, Headley JV . (2005). In situ bioremediation of naphthenic acids contaminated tailing pond waters in the Athabasca oil sands region--demonstrated field studies and plausible options: a review. J Environ Sci Health A Tox Hazard Subst Environ Eng 40: 685–722.
Ramos-Padrón E, Bordenave S, Lin S, Bhaskar IM, Dong X, Sensen CW et al. (2011). Carbon and sulfur cycling by microbial communities in a gypsum-treated oil sands tailings pond. Environ Sci Technol 45: 439–446.
Rooney RC, Bayley SE, Schindler DW . (2012). Oil sands mining and reclamation cause massive loss of peatland and stored carbon. Proc Natl Acad Sci USA 109: 4933–4937.
Semrau JD, DiSpirito AA, Yoon S . (2010). Methanotrophs and copper. FEMS Microbiol Rev 34: 496–531.
Semrau JD . (2011). Bioremediation via methanotrophy: overview of recent findings and suggestions for future research. Front Microbiol 2: 209.
Sharp CE, Stott MB, Dunfield PF . (2012). Detection of autotrophic verrucomicrobial methanotrophs in a geothermal environment using stable isotope probing. Front Microbiol 3: 303.
Solomon S, Qin D, Manning M, Chen Z, Marquis M, Avery KB et al. (2007) Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press: Cambridge, United Kingdom and New York, NY, USA.
Sommer DD, Delcher AL, Salzberg SL, Pop M . (2007). Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8: 64.
Stein LY, Klotz MG . (2011). Nitrifying and denitrifying pathways of methanotrophic bacteria. Biochem Soc Trans 39: 1826–1831.
Stoecker K, Bendinger B, Schöning B, Nielsen PH, Nielsen JL, Baranyi C et al. (2006). Cohn’s Crenothrix is a filamentous methane oxidizer with an unusual methane monooxygenase. Proc Natl Acad Sci USA 103: 2363–2367.
Stralis-Pavese N, Abell GC, Sessitsch A, Bodrossy L . (2011). Analysis of methanotroph community composition using a pmoA-based microbial diagnostic microarray. Nat Protoc 6: 609–624.
Surget-Groba Y, Montoya-Burgos JI . (2010). Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res 20: 1432–1440.
Swart NC, Weaver AJ . (2012). The Alberta oil sands and climate. Nature Clim Change 2: 134–136.
Tavormina PL, Ussler W, Orphan VJ . (2008). Planktonic and sediment-associated aerobic methanotrophs in two seep systems along the North American margin. Appl Environ Microbiol 74: 3985–3995.
Tavormina PL, Orphan VJ, Kalyuzhnaya MG, Jetten MSM, Klotz MG . (2011). A novel family of functional operons encoding methane/ammonia monooxygenase-related proteins in gammaproteobacterial methanotrophs. Environ Microbiol Rep 3: 91–100.
Trotsenko YA, Khmelenina VN . (2002). Biology of extremophilic and extremotolerant methanotrophs. Arch Microbiol 177: 123–131.
van Bodegom P, Stams F, Mollema L, Boeke S, Leffelaar P . (2001). Methane oxidation and the competition for oxygen in the rice rhizosphere. Appl Environ Microbiol 67: 3586–3597.
Vorob’ev AV, Baani M, Doronina NV, Brady AL, Liesack W, Dunfield PF et al. (2011). Methyloferula stellata gen. nov., sp. nov., an acidophilic, obligately methanotrophic bacterium that possesses only a soluble methane monooxygenase. Int J Syst Evol Microbiol 61: 2456–2463.
Vuilleumier SP, Chistoserdova L, Lee MC, Bringel FO, Al Lajus, Zhou Y et al. (2009). Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. Plos One 4: e5584.
Wagner D, Lipski A, Embacher A, Gattinger A . (2005). Methane fluxes in permafrost habitats of the Lena Delta: effects of microbial community structure and organic matter quality. Environ Microbiol 7: 1582–1592.
Ward N, Larsen O, Sakwa J, Bruseth L, Khouri H, Durkin AS et al. (2004). Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol 2: e303.
Willems A, Gillis M . (2001). Family IV. Comamonadaceae. In: Bergey DH, Boone DR, Garrity GM, Castenholz RW, Brenner DJ, Krieg NR, Staley JT (eds) Bergey’s Manual of Systematic Bacteriology: The Proteobacteria: Part C. The Alpha-, Beta-, Delta-, and Epsilonproteobacteria. Springer: Berlin, pp 686–688.
Acknowledgements
We thank the mining companies for the support of this research. Particular thanks go to Samantha Tavener and Joe Fournier for providing samples. This research was supported by Genome Canada and Genome Alberta (Hydrocarbon Metagenomics) and the Institute for Sustainable Energy, Environment and Economy (ISEEE). CES was supported by doctoral fellowships from NSERC and Alberta Innovates-Technology Futures. ALB was supported by a postdoctoral award from NSERC. DNA Sequence data sets related to this project have been submitted to the Sequence Read Archive under accession number SRP013946.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Saidi-Mehrabad, A., He, Z., Tamas, I. et al. Methanotrophic bacteria in oilsands tailings ponds of northern Alberta. ISME J 7, 908–921 (2013). https://doi.org/10.1038/ismej.2012.163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.163
Keywords
This article is cited by
-
Functional genome-centric view of the CO-driven anaerobic microbiome
The ISME Journal (2021)
-
Novel copper-containing membrane monooxygenases (CuMMOs) encoded by alkane-utilizing Betaproteobacteria
The ISME Journal (2020)
-
Large freshwater phages with the potential to augment aerobic methane oxidation
Nature Microbiology (2020)
-
A shared core microbiome in soda lakes separated by large distances
Nature Communications (2019)
-
DNA-SIP based genome-centric metagenomics identifies key long-chain fatty acid-degrading populations in anaerobic digesters with different feeding frequencies
The ISME Journal (2018)
