
Abstract
The bacteria that colonize the gastrointestinal tracts of mammals represent a highly selected microbiome that has a profound influence on human physiology by shaping the host’s metabolic and immune system activity. Despite the recent advances on the biological principles that underlie microbial symbiosis in the gut of mammals, mechanistic understanding of the contributions of the gut microbiome and how variations in the metabotypes are linked to the host health are obscure. Here, we mapped the entire metabolic potential of the gut microbiome based solely on metagenomics sequencing data derived from fecal samples of 124 Europeans (healthy, obese and with inflammatory bowel disease). Interestingly, three distinct clusters of individuals with high, medium and low metabolic potential were observed. By illustrating these results in the context of bacterial population, we concluded that the abundance of the Prevotella genera is a key factor indicating a low metabolic potential. These metagenome-based metabolic signatures were used to study the interaction networks between bacteria-specific metabolites and human proteins. We found that thirty-three such metabolites interact with disease-relevant protein complexes several of which are highly expressed in cells and tissues involved in the signaling and shaping of the adaptive immune system and associated with squamous cell carcinoma and bladder cancer. From this set of metabolites, eighteen are present in DrugBank providing evidence that we carry a natural pharmacy in our guts. Furthermore, we established connections between the systemic effects of non-antibiotic drugs and the gut microbiome of relevance to drug side effects and health-care solutions.
Similar content being viewed by others
Introduction
The adult human gastrointestinal tract is home to almost 1014 of bacteria, archaea, and eukarya cells, what is commonly referred to as the gut microbiome; thus it seems appropriate to view ourselves as a composite of many species. Our affiliated microbial partners contain ⩾100 times the number of genes embedded in our Homo sapiens genome, providing us with a genetic landscape and functional features we do not have to evolve on our own (Backhed et al., 2004). These tens of trillions of microbes that live in our gastrointestinal tract represent an anaerobic bioreactor programmed to synthesize several molecules that direct the human immune system locally and systematically (Hooper, 2004). The biosynthetic variability of our gut microbiota, which remains largely unexplored, could shape the human physiological phenotypes and is viewed as a modifier of the host epigenome (Li et al., 2008). As a matter of fact, the indigenous metabolic processes coded in the host metagenome are a source of pharmacologically active secondary metabolites involved in host metabolic regulation (Nicholson, 2006; Sonnenburg et al., 2006). Although we know that the symbiotic gut microbiome has evolved to exert control over a number of important mammalian metabolic regulatory functions, little is understood of the systems level interactions between the chemical signatures that characterize the organism-level metabolic status and host cellular pathways.
The first step in improving our understanding on how epigenetic or genetic changes that uncouple the shared evolutionary fate of humans and their symbiotic bacteria can result in disease, is to describe the composition of the metabolic network of microbial communities with respect to the healthy state. Turnbaugh et al. (2009) have shown that the gut microbial community structures of adult monozygotic twin pairs had a degree of similarity that was comparable to that of dizygotic twin pairs, and only slightly more similar compared with their mothers, whereas Palmer et al. (2007) revealed that gut community assembly during the first year of life followed a more similar pattern in a pair of dizygotic twins compared with unrelated infants. This interpersonal variation in the composition of the human microbiota implies that studying the role of gut bacteria in the development of pathophysiology and as complimentary metabolic machinery for drugs and diets across a small set of individuals may inappropriately treat these diverse phenomena as a single, albeit noisy phenomenon. In contrast, averaging the effects of a disturbance across large cohorts of individuals with distinct disease phenotypes can reveal the links between bacterial dynamics and host physiology and pathology.
These disturbances in a healthy adult microbiota can be the result of several factors, such as urbanization, diet or hygiene; however, there is a major concern that medical therapies may alter the composition of the human microbiota. Although antibiotic treatment is typically followed by a decrease in the diversity of the microbiota (Jernberg et al., 2007), it is unknown how other medical treatments might change the microbiota and deregulate the host immune homeostasis. Recent studies on drug polypharmacology (Chang et al., 2010; Ainsworth, 2011; Wang et al, 2011; Yabuuchi et al., 2011) indicate that many of the known drugs have more than one target and highlight the need to undertake a systems level approach to understand drug efficacy and adverse drug reactions considering the effect of a molecule in a global physiological environment, including the gut microbiota.
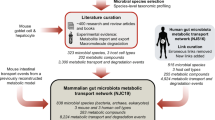
To get a deeper understanding of the microbial populations in the human digestive tract and their interactions with the host, we described the genetically defined metabotypes from fecal samples of 124 European adults (≈3.3 million genes). The cohort is divided into healthy and obese individual human adults, as well as inflammatory bowel disease (IBD) patients (Qin et al., 2010). Even though several studies have suggested that gut microflora composition is associated with obesity and IBD (Kau et al., 2011; Maloy and Powrie, 2011) the elucidation of the molecular mechanisms underlying microbe–host reciprocal metabolic interactions is far from complete. The goals of the present study were to (i) evaluate the variability in the metabolic potential of the gut microbiome in healthy individuals based on variations in the metabolic gene content of the bacterial population; (ii) apply a systems chemical biology approach to link the putative meta-metabolome space with the human protein and disease space and (iii) explore how medical treatments disturb the bacterial communities, how gut microbes affect drug metabolism and draw connections between side effects of drugs and alterations in human microbiota (Figure 1a).

The genetically defined metabotypes of the bacterial communities in the gut of healthy individuals. (a) The present study focuses on the evaluation of: (1) the interactions between small molecules (metabolites) produced by the microbiome and human proteins, and (2) the interactions between drug molecules and bacterial proteins. (b) The frequency of each of the 1490 metabolic reactions (red: essential reactions, blue: non-essential) that were assigned to the microbiome of the healthy individuals. The frequencies are given as both: (1) total number of reactions for a given frequency, represented by the bar width, and (2) density for essential and non-essential reactions for a given frequency. (c) Visualization of the most common (present in >70% of the samples) metabolic reactions (59 reactions) of the group of healthy individuals. Glycolysis, pyruvate metabolism, fatty acid metabolism, amino acid and nitrogen metabolism were the most conserved parts of the metabolic network. (d) The three distinct groups formed by clustering the healthy individuals based on a binary representation of their microbiome metabolic profile. The three clusters are characterized by high (736–1395), medium (254–758) and low (34–195) number of metabolic reactions. (e) Distribution of reaction counts in healthy samples dominated by the Bacteroides, Prevotella, and Ruminococcus genera (Arumugam et al., 2011).
Materials and methods
Metabolic proteins, reactions and metabolites
Enzymes, reactions and metabolite data were acquired from MetaCyc 14.6, a database that contains information about experimentally demonstrated small-molecule metabolic reactions and pathways, with links to sequences of the responsible enzymes (Caspi et al., 2010). MetaCyc contains a total of 9299 unique reactions, of which protein sequences could be retrieved for 6890. MetaCyc also contains 8869 metabolites, 8468 of which with known structure.
Human metagenome data
The metagenome data of the human microbiome are part of the Metagenomics of the Human Intestinal Tract (MetaHit) project (Qin et al., 2010). The data consist of gene sequences, distribution of genes among individuals (healthy samples=53, obese samples=46, IBD samples=10) and metadata for a subset of the samples, describing other non-genomic characteristics. The raw Illumina read data of all 124 samples have been deposited in EBI, under the accession ERA000116. The contigs and gene set are available to download from EMBL (http://www.bork.embl.de/~arumugam/Qin_et_al_2010/) and BGI (http://gutmeta.genomics.org.cn) websites.
Linking MetaHit with MetaCyc database
All nucleotide sequences from the MetaHit FASTA-files were given in their respective reading frame and could thus be translated directly to the amino acid sequences. The link between the MetaHit data and the MetaCyc database was initially established by finding homolog proteins using the BLAST-Like Alignment Tool (BLAT) (Kent, 2002). Two proteins were considered homologs if there was at least 80% coverage of the MetaHit protein sequence with 85% identity (amino acid level). A set of metabolic proteins and the respective metabolic network were assigned to each sample. For the metabolic proteins that were found in >70% of the samples, the KEGG IDs of the respective metabolic reactions were retrieved and visualized using iPATH (Yamada et al., 2011). The abundance per sample of each metabolic gene is not depicted in the iPATH visualization.
Species diversity
Taxonomical information regarding each MetaHit sequence aligned to a MetaCyc sequence was acquired by aligning all MetaHit sequences to sequences of known origin, using 90% identity threshold (nucleotide level), describing classes ranging from super kingdom to strain (Qin et al., 2010). Of the 3.3 million sequences of the non-redundant database, almost 500 000 were not directly associated to species.
Clustering and statistical analysis
The distance between samples was measured from their metabolic reaction profile using a binary matrix (‘1’: the reaction is present in an individual, ‘0’: the reaction is absent). The similarity between two samples was measured by the Tanimoto coefficient (Tc). The Tc gives a similarity value based on the overlap and size of the bit strings of the two samples under examination (Jonsdottir et al., 2005). Then the distance between samples is simply calculated as 1 minus Tc. The clustering of the samples was performed using hierarchical clustering with Ward’s method (Ward, 1963).
For each reaction, we calculated the mean abundance over all samples, where the reaction was present (for example, mean for healthy, obese and IBD). Subsequently, we calculated the log2-fold change between healthy and obese, and between healthy and IBD, respectively. We kept reactions with a log2-FC <−1 or >+1, and we ended up with 64 reactions for the obese samples and 128 for the IBD samples. We then performed a Wilcoxon rank test (data is sparse, and therefore t-tests are not suitable) on these reactions and P-values were adjusted using the false discovery rate (Benjamini–Hochberg).
Disease genes and complexes
Data on disease-relevant protein complexes were retrieved from our in-house database (Lage et al., 2008) (http://cbs.dtu.dk/cgi-bin/phecomplex/index.pl, last accessed on 31 August 2011), where complexes are linked to tissues through RNA expression data derived from 73 healthy tissues from the Novartis Research Foundation Gene Expression Database (www.gnf.org). A positive z-score indicates that the genes participating in a complex are expressed in the particular tissue at a higher level than the average gene of this tissue. The database consists of a list of 1524 protein complexes, assembled by 5202 unique proteins, connected through 45 662 unique interactions. The complexes are mapped to 1054 OMIM diseases. For 346 complexes the z-score for each of the 73 tissues is available. More information for the methodology on tissue specificity of disease complexes could be found in Lage et al. (2008).
Human metabolic network
In order to construct a subset of MetaCyc containing only non-human metabolites, human metabolomics data were acquired from the Homo sapiens Recon 1, a comprehensive literature-based genome-scale metabolic reconstruction of the global human metabolic map (Duarte et al., 2007). H. sapiens Recon 1 is part of the BIGG database (Schellenberger et al., 2010). All structures from MetaCyc and H. sapiens Recon 1 were first converted to Canonical SMILES using Open Babel (O’Boyle et al., 2011). SMILES (Simplified Molecular Input Line System) is a line notation for representing structures of small molecules. The generated SMILES is not unique for a given structure, thus, canonicalization involves the adoption of certain rules, which produce one generic SMILES among all valid possibilities (http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html). Metabolites with identical Canonical SMILES, which were present in both MetaCyc and H. sapiens Recon 1, were subsequently removed from the former and the remaining ones consisted the list of non-human metabolites.
Drugs and drug-target data
Small-molecule drug and target data were acquired from DrugBank (DB) v.3 (Knox et al., 2011) and the Therapeutic Target Database (TTD) 4.3.02 (Chen et al., 2009). DB contains information for 6708 drugs (including 1437 approved, 5086 experimental, 83 nutraceuticals and 134 protein/peptide drugs) linked to 4229 non-redundant targets. Small-molecule structures, as SMILES strings, were obtained for 6628 small molecules, while the sequences of the drug targets were retrieved as FASTA files (available for 4050 targets). The TTD contains information for 17 816 drugs (including approved, clinical, experimental and antisense drugs) linked to 2025 targets (including successful, clinical, discontinued and research targets). Structures were downloaded for 14 783 small molecules as MOL-files and were converted to SMILES strings using Open Babel. FASTA-files for 1502 protein targets were also available at the TTD website. After merging of the two datasets and removal of duplicates, the final list consisted of 19 917 unique small molecule structures and 4595 unique drug targets.
The similarity between the small molecule drugs and bacterial metabolites was accessed by the Tanimoto coefficient. Two molecules were considered similar when they fulfilled both the following criteria: (i) Tc⩾0.85, and (ii) at least one common protein interaction partner in ChEMBLdb.
Results
Metabolic individuality in the healthy human microbiome
To evaluate the metabolome profile of the gut of healthy individuals we defined the metabolic capacity of the bacterial populations by linking the MetaHIT protein sequences to the MetaCyc database (Caspi et al., 2010) for each individual. We could, in this manner, retrieve all the MetaHit genes (from now on called ‘metabolic genes’) coding for proteins that are part of a metabolic pathway (from now on called ‘metabolic proteins’). Using strict criteria, two proteins were considered to be homologs when they shared 85% identity and at least 80% coverage, and a total of 3775 MetaHIT sequences accounting for 1490 metabolic reactions were retrieved (Supplementary Table S1). We should emphasize here that the metabolome profile assigned to each individual is solely based on genomic data that indicate the metabolic potential of the bacterial population with no further evidence that the detected genes are actually expressed to produce the metabolites. A binary presence/absence (1/0) matrix was constructed using these 1490 metabolic reactions for the healthy samples. This matrix revealed a large variation in the total number of metabolic reactions that were assigned in the microbiome of each healthy individual. To investigate further this metabolic diversity in connection to the composition of the microbiome, we visualized the frequency of each metabolic reaction per individual taking into account its essentiality. Reactions were divided into essential and non-essential (Figure 1b) based on the Database of Essential Genes (Zhang and Lin, 2009) (DEG) 6.5 (http://essentialgene.org). By aligning the sequences from 18 prokaryotes in DEG to the MetaHIT sequences we retrieved 310 protein hits responsible for 425 essential metabolic reactions. Assigning a metabolic reaction as essential in our metagenome data should be always interpreted with caution so as not to neglect the symbiotic relationships in the bacterial population. Nevertheless from Figure 1b it is apparent that non-essential metabolic reactions have lower frequency among individuals compared with essential reactions (P-value <2.2 × 10−16 using Welch t-test). To capture a snapshot of the highly conserved among healthy individuals metabolic network, we visualized the metabolic reactions with frequency >70% using iPath2.0 (http://pathways.embl.de/). The parts of the microbiome metabolism that show a higher conservation among individuals are glycolysis and pyruvate metabolism, fatty acid metabolism, amino acid and nitrogen metabolism (Figure 1c).
Subsequently, the genetically defined metabotypes were visualized in a heat map (Figure 1d) where the distance between individuals is measured from their metabolic reaction profile using the binary matrix described above. Interestingly, three distinct clusters of healthy individuals with high, medium and low metabolic potential were observed. The larger group was the one with high metabolic potential, harboring between 736–1395 metabolic reactions. The number of reactions for the medium and low metabolic potential groups were 254–758 and 34–195, respectively. We have further confirmed that the clusters of metabolic potential are not the outcome of correlations with the general richness of the sample, or with sequencing depth (Supplementary File S1).
To illustrate these results in the context of bacterial population, we studied in what degree the variability in the metabolic potential of individuals is correlated with the abundance levels of the Bacteroides, Prevotella and Ruminococcus genera in each sample (Arumugam et al., 2011). If we attempt to draw direct connections between the abundance of the three genera and the richness of the metabolic network based on Figure 1e, a high correlation is observed only for the Prevotella genera; Prevotella shows strong presence in samples with low metabolic potential and it becomes virtually absent in samples with high number of metabolic reactions. However, we should keep in mind that our correlations between the three genera and metabolic potential are based solely on genomic data.
Generating connections of ‘non-human’ metabolites, protein complexes and diseases
To uncover potentially produced meta-metabolites that may have a significant contribution to health maintenance, we sought interactions between human proteins and bacterial metabolites that are not part of the human metabolic network. To create this set of metabolites we overlaid the putative gut microbiome metabolism with the Homo sapiens Recon 1 (Duarte et al., 2007) metabolic model. We obtained a set of 482 metabolites with known structure, which can be potentially synthesized or degraded only by the gut bacteria. This study revealed at the same time a significant overlap between the human and the bacterial metabolic capacity (376 shared metabolites), comparable to the number of human-specific metabolites (472 metabolites). The set of 482 metabolites that cannot be produced or metabolized by humans (‘non-human’ metabolites) was the base for the subsequent analysis. ChEMBLdb (Gaulton et al., 2011), a manually curated database of bioactive molecules, was used for generating an interactome map of the ‘non-human’ metabolites and the human proteome space. From the 62 metabolites that were linked to the human proteome (Supplementary Table S2), and after filtering for small organic molecules, 33 were found to be associated with a disease-relevant protein complex, while 18 of them were present in DrugBank (Knox et al., 2011) as experimental drugs. In the metabolic reactions of the metagenome, in which the ‘non-human’ metabolites are involved, five appear as substrates (S), eight appear as products (P), and the rest appear as both a substrate and a product (S and P) (Figure 2b). The taxonomy of genes encoding the bacterial enzymes involved in these reactions revealed Escherichia coli, Klebsiella pneumonia, Enterobacter cloacae (and E. cancerogenus), Shigella sp. and Citrobacter rodentium as the most efficient producers/consumers (Figure 2a). However, we should keep in mind that linking sequences with species was possible for only one-third of the enzymes.

The interactome space of the 33 ‘non-human’ metabolites with the human proteome. (a) A total of 195 MetaHit sequences (non-redundant) are involved in reactions in which these 33 metabolites participate. (1) Taxonomy distribution (top 20 species) for the MetaHit sequences (1091 non-redundant) involved in all metabolic reactions, (2) available taxonomy distribution for 56 of the 195 MetaHit sequences. (b) Heat map showing the disease space (given as OMIM IDs) targeted by the 33 metabolites that can be synthesized or degraded only by the gut microbiome. Information in parenthesis shows whether a metabolite participates in an irreversible reaction (as substrate (S) or a product (P)) or in a reversible reaction (S and P). When available, the Anatomical Therapeutically Classification code was retrieved from DrugBank. (c) The tissue specificity (given as a z-value) of the disease protein complexes that interact with metabolites with high binding affinity (Ki⩽1 μM). Hierarchical clustering was performed using Euclidean distances according to Ward’s method. Detailed information for the protein complex IDs is provided in Supplementary Table S4.
The interactions of the ‘non-human’ metabolites with disease complexes were established using an in-house database, which includes 2227 unique proteins that have been associated with a disease OMIM (Online Mendelian Inheritance in Man) ID through data mining (Lage et al., 2008; Figure 2b). Disease protein complexes involved in squamous cell carcinoma, Li-Fraumeni syndrome, lung cancer and bladder cancer were associated with >10 ‘non-human’ metabolites each. Drug metabolism disorders were also ranked highly in the list, as several metabolites interact with complexes that include enzymes of the cytochrome P450 superfamily involved in the metabolism of drugs and other xenobiotics (Supplementary Table S3). Metabolites quercetin and menadione that were found to interact with disease protein complexes associated with a large number of OMIM IDs, seem to have a significant role in the host-bacterial mutualism. Along with these two ‘non-human’ metabolites, hydroquinone and S-carboxylmethyl-D-cystein formed a cluster sharing many of the disease IDs. Quercetin, menadione and S-carboxymethyl-D-cysteine participate in reversible reactions offering a higher degree of flexibility for regulating their concentrations in the gut, whereas hydroquinone appears as product of an irreversible reaction, according to MetaCyc.
Figure 2c illustrates the tissue specificity (given as a z-value) for the disease complexes, where at least one member protein is targeted with high affinity by one of the ‘non-human’ metabolites (detailed information for the disease complex IDs is given in Supplementary Table S4). Five metabolites, namely quercetin, erythronate-4-phosphate, D-glucono-1,5-lactone, 5′,5″-diadenosine triphosphate and hydroquinone, were associated with disease complexes, because of experimental binding activity to a member of the complex of Ki⩽1 μM, which is a common threshold when considering efficient, small compound ligands (Overington et al., 2005). A cluster on the lower left part of this figure caught our attention, with 13 disease complexes related with different types of cancer that are highly expressed in cells and tissues involved in the signaling and shaping of the adaptive immune system (such as NK cells, B cells, dendritic cells, thymus, monocytes). Quercetin shows high affinity interactions with 11 of the complexes, whereas hydroquinone and D-glucono-1,5-lactone interact with one complex each. The protein complex targeted by hydroquinone is involved in DNA repair (GO:0006281, P-value⩽5.10e−15) supporting evidence that this metabolite acts as an inducer of cytogenetic changes and DNA hypomethylation (Ji et al., 2010), whereas the protein complex targeted by D-glucono-1,5-lactone is involved in transcription regulation (GO:0045449, P-value⩽5.29e−22).
Modulation of obesity and IBD by the gut microbiota
The MetaHIT cohort also contains samples from patients with IBD as well as individual fat percentage values for some samples, which can be used to group people as obese, allowing us to test if variations in the gut metabotypes have a role in the progression of these diseases. Following the same approach as the one used for the healthy individuals, we defined the metabolic network of the bacterial community for obese (1487 reactions) and IBD patients (1480 reactions). When clustering the samples for obese and IBD patients, based on binary data, we observed a similar trend as for the healthy individuals with three clusters of high, medium and low metabolic potential (data not shown). As the binary data could not reveal any significant differences in the metabolic potential between healthy and disease samples, we evaluated the abundance of the metabolic reactions per individual in each group (healthy, obese and IBD). The term abundance corresponds to the frequency of the sequences coding for each metabolic protein within the complete set of metabolic genes of the individual. In contrast to the binary data, attempting to cluster the samples of each group based on the abundance values resulted in no distinct clusters. Supplementary Figure S1 shows the most abundant metabolic reactions for healthy, obese and IBD samples. N-succinyl ornithine carbamoyl transferase and GDP-mannose 4,6 dehydratase were the most abundant metabolic genes not only in healthy but also in obese and IBD samples. When comparing the top-20 most abundant metabolic genes between the different groups very few differences were observed. In the comparison of the healthy against the other two groups, 3-aminobutyryl-CoA ammonia lyase, N-acetylhexosamine 1-kinase and formate C-acetyltransferase were the only reactions that did not appear in the top-20 of obese individuals, while pyruvate synthase, dimethylmaleate hydratase, acetyl-CoA-acetyl transferase, pyridoxal 5′-phosphate synthase (glutamine hydrolyzing) and hydrogensulfite reductase are the ones absent in the IBD top-20 most abundant metabolic genes.
In order to find statistically significant differences in the metabolic potential between healthy, obese and IBD samples, we applied a Wilcoxon signed-rank test, and used false discovery rate correction for multiple testing. The abundance of only two metabolic genes was found to be significantly different (P-value<0.05, both lower abundant in IBD individuals). The first reaction (E.C.1.3.5.4) is catalyzed by fumarate reductase, whereas the second one is the oxidation of menadiol by quinol monooxygenase yielding menadiole, superoxide and H+. As there is an increasing focus on oxidative stress as potential etiological factor and/or triggering factor in Crohn's disease (Iborra et al., 2011), it was interesting to find a reaction containing superoxide significantly different between healthy and IBD patients. While in the literature there are metabolomic-based studies performed in humans or model systems that have reported differences in the concentration of specific metabolites between healthy and non-healthy (obese, IBD) individuals (Waldram et al., 2009; Turnbaugh et al., 2010; Le Gall et al., 2011; Ponnusami et al., 2011), our analysis points out that these differences are not the result of genomic variability in the metabolic potential of the gut microflora. The analysis of transcripts would be the next natural step of the present study, in order to investigate whether there is a uniform functional pattern in healthy, obese and IBD samples or the differences in the metabolome level observed in other studies are the result of differences at the mRNA level.
Drug effects on host-microbiome mutualism
To identify drug treatments that are affected the most by the gut microbiome and vice versa, we followed two approaches: (i) we studied the chemical similarity between all drug molecules present in DrugBank and the metabolites of the metabolic network of healthy individuals. A drug and a compound were considered similar if they had a Tanimoto coefficient⩾0.85 and shared at least one known protein interactor in ChEMBLdb. (ii) We evaluated the sequence similarity (90% similarity and 80% coverage) of the drug targets with the metabolic and non-metabolic proteins of the metagenome of healthy individuals. In total, 603 experimental and approved drugs (among them 42 antibiotics) were predicted to perturb 515 unique metabolic reactions (64 drugs linked to metabolic reactions based on drug-metabolite chemical similarity and 556 drugs based on target homology). From these metabolic reactions, 161 are essential reactions (DEG 6.5) (Figure 3a). Interestingly, 16 experimental drugs and 5 approved drugs are identical to metabolites that act as substrates in the microbiome metabolic network, indicating that their therapeutic activity is potentially affected by bacterial metabolism. Looking into the non-metabolic targets of drugs, based on the sequence similarity of the drug target and the MetaHit sequences, we found 334 drugs (among them 60 antibiotics) that could potentially modulate the activity of 495 proteins (Figure 3b) of the bacterial community (170 essential proteins). From the above drug–microbiome interactions there were several notable cases. Tobramycin interacts with two non-metabolic proteins, which are both assigned as essential. Tobramycin is an antibiotic used to treat various types of bacterial infections and shows diarrhea and abdominal pain as highly common side effects (www.sideeffects.embl.de). Olanzapine and aripirpazol were shown to interact each with one non-metabolic protein of the gut metagenome. These two antipsychotic drugs show diarrhea, dyspepsia, constipation and gain weight (only olanzapine) as common side effects. Cytarabine, an anticancer drug, targets five metabolic proteins (one of them essential) of the bacterial metabolic network. According to SIDER constipation is a common side effect associated with this treatment. In Table 1, we present the 18 drugs and their respective Anatomical Therapeutic Chemical Classification codes with the most relevant, to the gut microbiome, side effects (diarrhea, flatulence and so on).

Drug–metagenome interactions. (a) A network of interactions between drug molecules (circles) and metabolic reactions (squares) in healthy individuals. A drug is connected with a metabolic reaction if (i) it is chemically similar with a metabolite that appears as reactant and (ii) the drug and the metabolite share at least one protein interactor in ChEMBLdb. Red font indicates essential reactions and green font indicates antibiotics. (b) A network of interactions between drug molecules (circles) and non-metabolic proteins (squares) in healthy individuals. A drug is connected with a protein if the drug target and the metagenome protein are homologs (90% similarity and 80% coverage). Red font indicates essential reactions and green font indicates antibiotics.
Discussion
The role of the human intestinal microbiome in metabolizing dietary constituents and protecting the host against pathogens is crucial to human health (Ley et al., 2008). Analysis of genomic and metagenomic sequences has produced gene catalogs with protein families involved in the predominant functions of the gut bacteria (Qin et al., 2010; Turnbaugh et al., 2010). However, currently lacking is a large-scale systematic analysis of the complex chemical space generated by these bacteria and the interaction with the human proteome. By defining the genetically defined metabotypes of >120 individuals, we concluded that independent of healthy or diseased samples, the individuals are clustered as high, medium or low metabolic potential. Further to this, Prevotella genera appear to be highly sensitive to the complexity of the metabolic chemical space.
A comparison of the metagenome metabolic network with the human metabolic network revealed a large number of metabolites that appear as reactants only in the bacterial metabolism. The finding that from this set of metabolites, 18 are present in DrugBank provides evidence that we carry a natural pharmacy in our guts. In addition, 15 other ‘non-human’ metabolites were found to interact with a set of human protein complexes associated with a wide spectrum of diseases. For example quercetin, a flavonoid compound that is widely distributed in nature, and menadione, a vitamin precursor of K2, were found to interact with disease protein complexes associated with a large number of OMIM IDs. Although there are no official health claims granted to quercetin, several studies have been conducted that report reduction of systolic blood pressure and plasma oxidized low-density lipoprotein concentrations in overweight subjects with a high cardiovascular disease risk phenotype (Egert et al., 2009), potential anti-obesity effects (Yang et al., 2008) and protection from colon and prostate cancer (Senthilkumar et al., 2011; Bae et al., 2012). Using here compound-protein interaction data we associated quercetin with disease protein complexes involved in both obesity and IBD. Menadione is a clinically important chemotherapeutic agent used in the treatment of leukemia and other types of cancer (Laux and Nel, 2001; Matzno et al., 2008). Due to adverse effects associated with it, menadione is used as nutritional supplement only in few developing countries, but is still used in many countries as inexpensive micronutrient for livestock. Understanding the regulation of the metabolic pathways in which the above two and the rest of the 33 metabolites are involved, and monitoring their actual concentration in healthy and disease individuals, could guide us to rationally design diets that promote host homeostasis. However, as genomic data provide only a blueprint or inventory of genes encoding enzymes without any proof of functionality, meta-transcriptomic or meta-metabolomic analysis is necessary before we attempt to shape the bacterial metabolic network activity to benefit the host. Especially, the power of meta-metabolomic analysis for establishing direct links between alterations in the gut microbiome and human host responses has been demonstrated in several studies. Martin et al. (2010) applied microbial profiling of fecal contents in four groups of animals to study dietary-specific modulation of microbial populations and a different profile of amino acids and SCFAs was observed upon modification of the dietary pattern (glucose–lactose mix) in conventional and non-conventional animals. In another notable study Yap et al. (2008) were the first to show that major perturbation of microbiome metabolites manifests with changes to the host’s systemic metabolic phenotype.
Antibiotics have been used effectively for over half a century to treat bacterial infections; however, it is widely accepted that they can also markedly affect the composition of the microbiota. Although the effect of antibiotics is short term, as typically most families and genera of gut microorganisms return to typical levels within weeks of exposure, there is no information on how the bacterial communities are affected by other classes of drugs, especially drugs with long-term use from the human host. From the computational analysis applied in our study there are indications that antibiotics are not the only therapeutic treatment that can result in a dysregulation of the symbiosis between the host and its microbiota. Antipsychotic and anticancer drugs were also found to potentially interact with bacterial proteins, and these interactions might explain observed side effects, such as diarrhea and constipation; however, experimental evidence is required to validate these observations. Zheng et al. (2011) have determined the global metabolic effects and dynamic changes of 223 fecal metabolites in antibiotic-treated rats and revealed alterations in metabolic systems not previously correlated with antibiotics. In parallel, Jakobsson et al. (2010) analyzed the 16S rRNA gene using 454-based pyrosequencing and terminal-restriction fragment length polymorphism of bacterial populations in human fecal samples after antibiotic administration and revealed marked ecological disturbances that naturally influence the abundance of specific metabolites. These are just a few of the experimental approaches that could be used for studying experimentally drug–microbiome interactions. In contrast to antibiotics, antipsychotic and anticancer drugs are long-duration treatments; therefore a mechanistic understanding of their side effects is of utmost importance.
References
Ainsworth C . (2011). Networking for new drugs. Nat Med 17: 1166–1168.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR et al. (2011). Enterotypes of the human gut microbiome. Nature 473: 174–180.
Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101: 15718–15723.
Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JL . (2005). Host-bacterial mutualism in the human intestine. Science 307: 1915–1920.
Bae JH, Kim SJ, Kim MJ, Oh SO, Chung JS, Kim SH et al. (2012). The susceptibility to NK cell-mediated lysis of colon cancer cells is enhanced by treatment with EGFR inhibitors through induction of ULBP1. Cancer Sci 103: 7–16.
Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F et al. (2010). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 38: D473–D479.
Chang RL, Xie L, Xie L, Bourne PE, Palsson BO . (2010). Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLoS Comput Biol 6: e1000938.
Chen X, Ji ZL, Chen YZ . (2009). TTD: therapeutic target database. Nucleic Acids Res 9: 197–201.
Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD et al. (2007). Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA 104: 1777–1782.
Egert S, Bosy-Westphal A, Seiberl J, Kürbitz C, Settler U, Plachta-Danielzik S et al. (2009). Quercetin reduces systolic blood pressure and plasma oxidized low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: A double-blinded, placebo-controlled cross-over study. Br J Nutr 102: 1065–1074.
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A et al. (2011). ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40: (Database issue) D1100–D1107.
Hooper LV . (2004). Bacterial contributions to mammalian gut development. Trends Microbiol 12: 129–134.
Iborra M, Moret I, Rausell F, Bastida G, Aguas M, Cerrillo E et al. (2011). Role of oxidative stress and antioxidant enzymes in Crohn's disease. Biochem Societ Trans 39: 1102–1106.
Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L . (2010). Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One 5: e9836.
Jernberg C, Lofmark S, Edlund C, Jansson JK . (2007). Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J 1: 56–66.
Ji Z, Zhang L, Peng V, Ren X, McHale CM, Smith MT et al. (2010). A comparison of the cytogenetic alterations and global DNA hypomethylation induced by the benzene metabolite, hydroquinone, with those induced by melphalan and etoposide. Leukemia 24: 986–991.
Jonsdottir SO, Jorgensen FS, Brunak. S . (2005). Prediction methods and databases within chemoinfromatics: emphasis on drugs and drug candidates. Bioinformatics 21: 2145–2160.
Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI . (2011). Human nutrition, the gut microbiome and the immune system. Nature 474: 327–336.
Kent S . (2002). BLAT-The BLAST-like alignment tool. Genome Res 12: 656–664.
Knox CV, Law V, Jewison T, Liu P, Ly S, Frolkis A et al. (2011). DrugBank 3.0: a comprehensive resource for “omics” research on drugs. Nucleic Acids Res 39: D1035–D1041 (database issue).
Lage K, Hansen NT, Karlberg EO, Eklund AC, Roque FS, Donahoe PK et al. (2008). A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci USA 105: 20870–20875.
Laux I, Nel A . (2001). Evidence that oxidative stress-induced apoptosis by menadione involves Fas-dependent and Fas-independent pathways. Clin Immunol 101: 335–344.
Le Gall G, Noor SO, Ridgway K, Scovell L, Jamieson C, Johnson IT et al. (2011). Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res 10: 4208–4218.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS et al. (2008). Evolution of mammals and their gut microbes. Science 320: 1647–1651.
Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H et al. (2008). Symbiotic gut microbes modulate human metabolic phenotypes. PNAS 105: 2117–2122.
Maloy KJ, Powrie F . (2011). Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474: 298–306.
Martin FPJ, Sprenger N, Montoliu I, Rezzi S, Kochhar S, Nicholson JK . (2010). Dietary modulation of gut functional ecology studied by fecal metabolomics. J Proteome Res 9: 5284–5295.
Matzno S, Yamaguchi Y, Akiyoshi T, Nakabayashi T, Matsuyama K . (2008). An attempt to evaluate the effect of vitamin K3 using an enhancer of anticancer agents. Biol Pharm Bull 31: 1270–1273.
Nicholson JK . (2006). Global systems biology, personalized medicine and molecular epidemiology. Mol Syst Biol 2: 52.
O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR . (2011). Open Babel: An open chemical toolbox. J Cheminform 3: 33.
Overington JP, Al-Lazikani B, Hopkins AL . (2005). How many drug targets are there? Nat Reviews 5: 993–996.
Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO . (2007). Development of the human infant intestinal microbiota. PLoS Biol 5: e177.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al. (2010). A human gut microbial gene catalogue established by metagenomics sequencing. Nature 464: 59–65.
Schellenberger J, Park JO, Conrad TM, Palsson BO . (2010). Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics. 11: 213.
Senthilkumar K, Arunkumar R, Elumalai P, Sharmila G, Gunadharini DN, Banudevi S et al. (2011). Quercetin inhibits invasion, migration and signaling molecules involved in cell survival and proliferation of prostate cancer cell line (PC-3). Cell Biochem Funct 29: 87–95.
Sonnenburg JL, Chen CT, Gordon JI . (2006). Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol 4: e413.
Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F et al. (2010). Organismal, genetic and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc. Natl. Acad. Sci USA 107: 7503–7508.
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE et al. (2009). A core gut microbiome in obese and lean twins. Nature 457: 480–484.
Waldram A, Holmes E, Wang Y, Rantalainen M, Wilson ID, Tuohy KM et al. (2009). Top-down systems biology modeling of host metabotype-microbiome associations in obese rodents. J Proteome Res 8: 2361–2375.
Wang H, Ding Y, Tang J, Dong X, He B, Qiu J et al. Finding complex biological relationships in recent pubmed articles using Bio-LDA. PLoS One 6: e17243.
Ward JH . (1963). Hierarchical grouping to ptimize an objective function. J Amer Statist Assoc 58: 236–244.
Yabuuchi H, Niijima S, Takematsu H, Ida T, Hirokawa T, Hara T et al. (2011). Analysis of multiple compound–protein interactions reveals novel bioactive molecules. Mol Syst Biol 7: 472.
Yamada T, Letunic S, Okuda M, Bork P . (2011). iPATH2.0: interactive pathway explorer. Nucleic Acids Res 39: 1–4.
Yang JY, Della-Fera MA, Rayalam S, Ambati S, Hartzell DL, Park HJ et al. (2008). Enhanced inhibition of adipogenesis and induction of apoptosis in 3T3-L1 adipocytes with combinations of reservatol and quercetin. Life Sci 82: 1032–1039.
Yap. IKS, Li JV, Saric J, Martin FP, Davies H, Wang Y et al. (2008). Metabonomic and microbiological analysis of the dynamic effect on vancomycin-induced microbiota modification in the mouse. J Proteome Res 7: 3718–3728.
Zhang R, Lin Y . (2009). DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res 37: D455–D458 (database issue).
Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NH et al. (2011). The footprints of gut microbial-mammalian co-metabolism. J Proteome Res 10: 5512–5522.
Acknowledgements
We would like to thank MZ Khodaie and K Jensen for valuable input in the statistical and computational analysis of the data. We would also like to thank Dr N Blom for fruitful discussions and Dr A Eklund for proofreading the paper. Finally, we would like to thank the Novo Nordisk Foundation for financial support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Jacobsen, U., Nielsen, H., Hildebrand, F. et al. The chemical interactome space between the human host and the genetically defined gut metabotypes. ISME J 7, 730–742 (2013). https://doi.org/10.1038/ismej.2012.141
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.141
Keywords
This article is cited by
-
The human microbiome, from Achilles armour to Nessus’ shirt
Archives of Toxicology (2017)
