
Abstract
As obligate symbionts of most land plants, arbuscular mycorrhizal fungi (AMF) have a crucial role in ecosystems, but to date, in the absence of genomic data, their adaptive biology remains elusive. In addition, endobacteria are found in their cytoplasm, the role of which is unknown. In order to investigate the function of the Gram-negative Candidatus Glomeribacter gigasporarum, an endobacterium of the AMF Gigaspora margarita, we sequenced its genome, leading to an ∼1.72-Mb assembly. Phylogenetic analyses placed Ca. G. gigasporarum in the Burkholderiaceae whereas metabolic network analyses clustered it with insect endobacteria. This positioning of Ca. G. gigasporarum among different bacterial classes reveals that it has undergone convergent evolution to adapt itself to intracellular lifestyle. The genome annotation of this mycorrhizal-fungal endobacterium has revealed an unexpected genetic mosaic where typical determinants of symbiotic, pathogenic and free-living bacteria are integrated in a reduced genome. Ca. G. gigasporarum is an aerobic microbe that depends on its host for carbon, phosphorus and nitrogen supply; it also expresses type II and type III secretion systems and synthesizes vitamin B12, antibiotics- and toxin-resistance molecules, which may contribute to the fungal host's ecological fitness. Ca. G. gigasporarum has an extreme dependence on its host for nutrients and energy, whereas the fungal host is itself an obligate biotroph that relies on a photosynthetic plant. Our work represents the first step towards unraveling a complex network of interphylum interactions, which is expected to have a previously unrecognized ecological impact.
Similar content being viewed by others
Introduction
Arbuscular mycorrhizal fungi (AMF, phylum Glomeromycota) are soil fungi that form a symbiotic association with most land plants (Smith and Read, 2008). This association dates back to the early Devonian times and is now widespread in all ecosystems (Fitter et al., 2011). The success of this symbiosis, both over time and in geographical distribution, is due to the benefits that both partners gain from the reciprocal nutrient exchange. AMF are known to possess endobacteria in their cytoplasm, which were first recognized on the basis of electron microscopy studies (Bonfante and Anca, 2009). Unlike insect endosymbionts, which are localized in specialized tissues (Moran et al., 2008), endobacteria are found in both extra- and intraradical hyphae, the latter colonizing the plant tissues. On the basis of their morphological features, two AMF endobacterial morphotypes can be distinguished. The first is coccoid in shape and is present in the cytoplasm of most AMF: these endobacteria have been phylogenetically related to Mollicutes (Naumann et al., 2010). The second type of endobacteria is restricted to the Gigasporaceae AMF family, is rod-shaped, is related to Burkholderia and is vertically transmitted (Bianciotto et al., 2004). These endobacteria are separated from the fungal cytoplasm by a membrane of fungal origin, and have been described as an uncultured taxon, which was named Candidatus Glomeribacter gigasporarum (Bianciotto et al., 2003). Studies on a fungal line from which these endobacteria had been removed demonstrated that Ca. G. gigasporarum contributes to the fitness of its host by improving the expansion and branching of pre-symbiotic hyphae (Lumini et al., 2007).
Although bacterial endosymbionts of insects, worms, sponges, amoebae and plants have been investigated in depth, and the genome sequence is available for many of them, the genomic characterization of fungal endobacteria is limited to a single isolate (Lackner et al., 2011a), and their biological significance is largely unknown.
To understand the metabolic processes underpinning the interaction of Ca. G with its fungal host, we sequenced the genome of a homogeneous bacterial population thriving in the BEG 34 strain of Gigaspora margarita, an AMF that in turn is an obligate plant biotroph. The Ca. G. gigasporarum genome was found to contain a mosaic of genetic determinants reminiscent of symbiotic, saprotrophic and pathogenic species. Analysis of its coding capacity revealed a strong metabolic dependence on the fungal host, thus confirming Ca. G. gigasporarum as an unculturable microbe. As AMF depend on the supply of carbon from the plant, Ca. G. gigasporarum is also ultimately dependent on the plant, creating a network of previously unrecognized ecological interactions in the soil.
Materials and methods
Biological materials
G. margarita Becker and Hall spores (strain BEG34; European Bank of Glomeromycota) were used for all the experiments. Spores are referred to as a wild type if they harbor endobacteria (B+), or as ‘cured’ (B−) if they do not contain endobacteria (Lumini et al., 2007). The biological material was processed as described in Supplementary Text S1.
Genomic DNA source and library construction
Two complementary strategies were used to sequence the Ca. G. gigasporarum genome: (1) Sanger sequencing via transposon tagging of fosmid clones selected from a metagenomic fosmid library and (2) shotgun 454 pyrosequencing. The metagenomic fosmid library was constructed from 8000 B+ G. margarita BEG34 spores; total genomic DNA was extracted according to a modified Moller et al. protocol (1992), and purified using QIAGEN Genomic Tip 100/G. The fosmid library was created with the Copy Control Fosmid Kit (Epicentre Biotechnologies, Madison, WI, USA), following the JGI Fosmid (40 kb) Library Creation Protocol v.2.1. The resulting library contained 36 000 primary recombinant clones with an average insert size of 35 kbp.
Bacterial isolation, DNA extraction and whole-genome amplification (WGA) for the pyrosequencing were performed as described in Supplementary Text S1.
Sequencing and assembly strategies
The detailed sequencing strategies are illustrated in Supplementary Text S1.
Sanger sequencing
Around 1098 fosmid primary clones were purified and the terminal fosmid ends were sequenced. A total of 2244 sequences were obtained, processed for quality and analyzed to search for similarity in public databases. Clones showing similarities with Burkholderiaceae were validated by PCR. All the experiments were performed using B+ G. margarita BEG34 DNA as the template and B− DNA as the negative control. A total of 68 validated clones were sequenced using a transposon-tagging approach. Subclones were processed and sequenced, yielding a total of 14 949 sequences.
454-Pyrosequencing
Only WGA-DNAs showing less than 7% fungal contamination were sequenced (Supplementary Text S1). A single sequencing run was performed by ROCHE (Branford, FL, USA) using GS FLX. A total of 442 958 high-quality filtered sequence reads were generated with a total sequence output of 106 336 407 bp. The multistep approach used to build the solid hybrid assembly and eliminate fungal contamination and the chimeric reads is described in detail in Supplementary Text S1. The final assembly included 125 contigs with a total length of 1 726 950 bp.
The Ca. G. gigasporarum nucleotide sequence and the annotation have been deposited in the EMBL Nucleotide Sequence Database under the following accession numbers: CAFB01000001–CAFB01000124.
Annotation and metabolic reconstruction
Automatic gene prediction was performed using the AMIGene software (Bocs et al., 2003). A total of 2058 coding sequences (CDSs) were predicted and subjected to automatic functional annotation (Vallenet et al., 2006). Putative orthologs and groups of orthologs between Ca. G. gigasporarum and all the other complete bacterial genomes (RefSeq) were computed on the MaGe platform as described in Vallenet et al. (2006). All the CDSs were manually inspected and validated. Four rounds of prediction and validation were used to produce the final version (v.4) of the Ca. G. gigasporarum genome annotation. All the data were stored in an instance of the Genome Annotations Management System MicroScope (Vallenet et al., 2009) called GlomeriScope (http://www.genoscope.cns.fr/agc/microscope/).
Two automatic pathway depictions, generated by KEGG (Kanehisa et al., 2004) and MicroCyc, based on MetaCyc (Caspi et al., 2010), were compared with each other and manually verified to assess false positive and negatives. For details see Supplementary Text S1.
Hierarchical cluster analysis of KEGG-predicted metabolic pathways for Ca. G. gigasporarum and other 28 genomes integrated in the PkGDB database was performed with MeV v.4.2 (http://www.tm4.org/mev/). Analysis of hierarchical trees of gene clusters has been performed using the Self-Organizing Tree Algorithm with default parameters.
Phylogenetic analyses
Three different analyses were performed to validate previous phylogenetic placement of Ca. G. gigasporarum (Lumini et al., 2006; Castillo and Pawlowska, 2010). First, 21 structural genes retrieved from 67 completely sequenced bacterial species were used for a large multigene phylogenetic analysis (Supplementary Table S1). As a further step, the analysis was restricted to β-proteobacteria, whose both 16S and 23S were available (Supplementary Table S2 and Supplementary Text S1).
Gene expression analysis
RNA extraction and real-time reverse transcriptase-polymerase chain reaction analysis were performed according to Anca et al. (2009). For details see Supplementary Text S1.
Results and Discussion
General features of Ca. G. gigasporarum genome
The assembly from Sanger sequencing and 454 pyrosequencing data sets revealed a genome size of 1 726 950 bp (N50: 49.770 bp; N90: 4467 bp). This value is in agreement with a previous estimation based on pulsed field gel electrophoresis (PFGE) analysis (Jargeat et al., 2004). The present genome assembly also includes all the sequence data previously obtained for Ca. G. gigasporarum on rRNAs, protein coding genes and expressed proteins (Ruiz-Lozano and Bonfante, 2000; Salvioli et al., 2008, 2010; Anca et al., 2009). These lines of evidence indicate that essentially all the Ca. G. gigasporarum genome has been sequenced.
The genome sequence was produced from 34 hybrid contigs, reconstructed from 68 Sanger-sequenced fosmid clones and shotgun 454 data, giving a total of 1 416 122 bp, and from an additional 91 contigs (310 828 bp), which were assembled only from 454 data (Supplementary Text S2).
Annotation of the Ca. G. gigasporarum genome provided evidence that it is organized into four genomic units: a chromosome and three plasmids (Supplementary Table S3 and Supplementary Text S2). In general, for endosymbionts, the smaller the genome the lower the GC content, whereas Ca. G. gigasporarum has a relatively high GC content (54.82%). This value is comparable to that of some other endosymbionts, for example, Ca. Hodgkinia cicadicola, which has one of the smallest known genome (144 kb), but a GC content of 58% (McCutcheon and Moran, 2010).
A total of 1884 genomic features were identified and annotated, including 1736 CDSs and 41 pseudogenes. Approximately 6.98% of the CDS products did not show similarity with known proteins. Both the average CDS size (867 bp) and the CDS density (75.88%) are lower than observed for other members of the Burkholderia genus. About 15% of the Ca. G. gigasporarum genome is constituted by repetitive elements, mainly including insertion sequences (IS) (Supplementary Table S4). The relatively low occurrence of active IS elements and pseudogenes suggests a reduced rate of evolutionary shuffling and an efficient removal of pseudogenes during evolution, as for other bacterial genomes (Kuo and Ochman, 2010). These events lead to a relatively stable genome structure suggesting an ancient origin of the interaction with its fungal host (Castillo and Pawlowska, 2010).
The Ca. G. gigasporarum genome codes for 38 tRNAs and one cluster of rRNA genes, in analogy to other strict endosymbionts, which often have only one copy of the ribosomal genes (Supplementary Text S2).
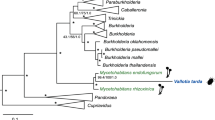
Phylogenetic inference was made on the basis of a multigene analysis, carried out on 21 proteins retrieved from 67 completed genomes belonging to a wide range of bacterial lineages (Supplementary Table S1). It indicated that Ca. G. gigasporarum belongs to the β-proteobacteria class, clustering within the Burkholderiaceae family (Figure 1), whose members are known to be highly versatile microbes interacting with animals, humans, plants and fungi (Bontemps et al., 2010; Hoffman and Arnold, 2010). B. rhizoxinica HKI 454, a fungal endobacterium, turned out to be the closest member, even though revealing a large genetic distance. Analysis of a data set restricted to β-proteobacteria (Supplementary Table S2) and based on 16S and 23S rRNA genes placed Ca. G. gigasporarum as a sister group of the Burkholderia clade, which includes free-living and other fungal-associated species (Supplementary Figures S1 and S2). These results suggest that Ca. G. gigasporarum is an ancient member of the taxon sharing a common ancestor with the present-day Burkholderiaceae.

Phylogenetic placement of CaGg among Eubacteria, inferred by multigene comparative maximum likelihood analysis, using 21 homologs from 67 completely sequenced genomes; the analysis revealed that CaGg is related to a large group of free-living Burkholderiaceae (β-proteobacteria; Burkholderiales). The 21 genes used for the phylogenetic analysis are listed in Supplementary Table S1. Bootstrap values >70% are indicated (1000 replicates). Kosmotoga olearia TBF 19.5.1 and Thermotoga lettingae TMO (Thermotogae; Thermotogales) were used as outgroup taxa to root the tree. *Taxa clustering together by comparative metabolic profile analysis (Figure 2). Scale bar, substitutions per site.
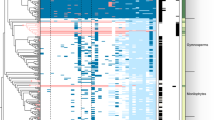
A hierarchical clustering analysis, based on the computation of ‘pathway completion’ (that is, the number of reactions for a pathway present in a given organism/total number of reactions in the same pathway defined in the KEGG databases), was applied to Ca. G. gigasporarum and other 28 bacterial genomes (Figure 2). The low completion values of the metabolic pathways required for amino-acid synthesis, fermentative and degradative capabilities, and glycolysis grouped Ca. G. gigasporarum to 13 insect endosymbionts, including Lawsonia intracellularis, Baumannia cicadellinicola, Ca. Hamiltonella defensa and Wolbachia spp. Interestingly, B. rhizoxinica is clustered with other cultivable β-proteobacteria. This finding indicates that genomes of endobacteria of phylogenetically distinct lineages have been shaped in a similar way by the selective pressures generated within diverse eukaryotic hosts and their specific environments. These data provide evidence of convergent evolutionary adaptation to an intracellular life style.

Detail of the hierarchical clustering of KEGG metabolic pathways predicted for CaGg and other 28 genomes integrated in the MaGe database. CaGg metabolic profile clusters with 13 sequenced genomes of intracellular endosymbionts, irrespectively of their far phylogenetic position, and not with the related fungal endosymbiont, Burkholderia rhizoxinica. Red box: 100% pathway completion; Green box: 0.0% completion. For the complete figure see Supplementary Figure S3 and the corresponding legend in Supplementary Text S4.
Predicted metabolic pathways
Little is known about the metabolic machinery of Ca. G. gigasporarum, as all attempts to cultivate the organism in vitro in a cell-free medium have been unsuccessful (Jargeat et al., 2004). Metabolic and functional analyses based on the Ca. G. gigasporarum genome annotation revealed interesting features related to the carbon flux (Figure 3). First, Ca. G. gigasporarum is restricted to an aerobic environment, as genes involved in fermentative pathways are not present in its genome. Its capability to obtain energy from complex sugars appears to be limited, as no starch- or sucrose-degradation enzymes were identified. Second, the glycolysis pathway is only partially represented, as the key glycolytic enzyme, phosphofructokinase, was not identified, which is also the case for the genome of the tsetse fly endobacterium, Wigglesworthia glossinidia (Akman et al., 2002), and that of an amoeba symbiont (Schmitz-Esser et al., 2010). In contrast, the gluconeogenesis pathway is complete.

Predicted reconstruction of the main metabolic pathways in CaGg. White circles represent enzymes whose corresponding genes have not been found in the genome for a given pathway. Partly represented pathways are not illustrated. arcD, arginine/ornithine antiporter; CYT, cytochrome C; DPP, dipeptide transporter; F0/F1, ATP synthase; MFS, major facilitator superfamily transporters; NDH-1, NAD dehydrogenase; POT, putrescine transporter; PST, phosphate transporter; PTS, sugar transporter, SDH, succinate dehydrogenase; TAT, twin-Arginine translocation pathway; TCA, tricarboxylic acid cycle.
In the context of the relatively complex carbon metabolism of Ca. G. gigasporarum(that is, limited capacity to gain energy via glycolysis, but sugar synthesis capacities), only a single copy of a sugar phosphate transporter gene (pts) was identified. By contrast, primary endosymbionts such as Buchnera, which lack gluconeogenesis capability, are fully sugar-dependent on their host, and consistently have multiple copies of the pts sugar transporter (Zientz et al., 2004).
The analysis of the metabolic capacity of Ca. G. gigasporarum strongly suggests that the organism is unable to synthesize the essential amino acids arginine, isoleucine, leucine, methionine, phenylalanine, tryptophan, histidine and valine. Aspartate, glutamate, glutamine, glycine, homoserine and threonine seem to be the only amino acids that can be de novo synthesized by Ca. G. gigasporarum (Supplementary Text S3). This reduced capability for amino-acid biosynthesis is similar to that of Wolbachia (Foster et al., 2005), but differs from that of other endosymbionts, such as Buchnera and Sulcia (McCutcheon and Moran, 2010). The identification of two genes for amino-acid permeases, two others for proteins belonging to the amino-acid carrier family, as well as fourteen CDS coding for the major facilitator super-family (MFS) with a putative specificity for amino-acid transport, provides clear evidence of amino-acid uptake by Ca. G. gigasporarum from its host.
As hypothesized for other endobacteria with a limited sugar metabolism (Wu et al., 2004), amino-acid catabolism is likely to represent a crucial source of energy for Ca. G. gigasporarum. Serine, glutamate, aspartate and arginine can in fact be catabolized with the production of energy. The complete arc operon, necessary for arginine import and catabolism, is indeed present in the Ca. G. gigasporarum genome. This system, called ‘the arginine deiminase pathway’, has also been described in the opportunistic species, Mycoplasma hominis, as a powerful way to generate ATP in bacteria with reduced metabolic capabilities (Pereyre et al., 2009). To verify whether the arc operon was active, quantitative reverse transcriptase polymerase chain reaction experiments were performed using the presymbiotic (spores and germinating spores) as well as the symbiotic (colonized roots) phases of the AMF life cycle. Transcripts for the arcA and arcB genes were detected in all the RNA samples tested, being significantly more abundant in germinating spores and when the fungal host was associated to the plant (Supplementary Figure S4). As arginine has a major role in the translocation of nitrogen from AMF to the host plant (Tian et al., 2010), it is likely that fungal-produced arginine is also used by Ca. G. gigasporarum to produce ATP.
Ca. G. gigasporarum has the whole genetic repertoire for nucleotide biosynthesis, and, in agreement with its rod-shape morphology and the layered Gram-negative wall, it possesses the genetic equipment required for lipopolysaccharide and peptidoglycan biosynthesis (Supplementary Table S5 and Supplementary Text S3). All the genes for fatty-acid and phospholipid biosynthesis were identified, while no evidence of beta-oxidation was found, suggesting that Ca. G. gigasporarum does not use fatty acids as a source of energy.
Ca. G. gigasporarum retains the pathways required for the biosynthesis of folate from dihydroneopterin and coenzyme A from pantothenate. However, no genes for pantothenate (vitamin B5), biotin (vitamin B7), thiamine (vitamin B1) or riboflavin (vitamin B2) biosynthesis were found, suggesting that these molecules may be provided by the fungal host.
A striking feature of the Ca. G. gigasporarum genome is the presence of 18 genes with similarity to those devoted to the synthesis of cobalamin (vitamin B12) from uroporphyrinogen III (Supplementary Table S5). Evidence for a functional vitamin operon is given by the detection of transcripts for three genes (cobN, cobI and cobQ) in quiescent and germinating G. margarita spores (Supplementary Figure S4). Cobalamin is produced via one of the most complex and exclusive prokaryotic pathways and has an essential role as a cofactor for several enzymatic reactions in animals, protists and prokaryotes. Among the endosymbionts, Ca. H. cicadicola, the co-habitant bacterium of Sulcia muelleri inside cicades, is so far the only endosymbiont known to have maintained the complete pathway (McCutcheon et al., 2009). The presence of a cobalamin biosynthetic pathway in a bacterial genome is often associated with the presence of cobalamin-dependent methionine synthase (metH) (McCutcheon et al., 2009). Surprisingly, neither cobalamin-independent nor cobalamine-dependent methionine synthase was detected in the Ca. G. gigasporarum genome, as the entire pathway for methionine biosynthesis seems to be missing. In the absence of an AMF genome sequence, it is still unknown whether the fungal host requires the vitamin B12 produced by Ca. G. gigasporarum. Interestingly, some algae have been shown to obtain vitamin B12 to support methionine synthesis through symbiotic associations with bacteria (Croft et al., 2005). Moreover, the essential role of vitamin B12 in plant–bacterial symbiosis has recently been demonstrated for the nitrogen-fixing Sinorhizobium meliloti, where the presence of a cobalamin-dependent ribonucleotide reductase was shown to be a central factor for the establishment of a symbiotic relationship with the host plant Medicago sativa (Taga and Walker, 2010).
In summary, the data obtained by the metabolic inference suggest that Ca. G. gigasporarum may generate energy from the degradation of amino acids taken up from its fungal host. Its capacity to synthesize sugars and fatty acids, as well as cofactors and vitamins, such as vitamin B12, indicates that Ca. G. gigasporarum is partially autonomous in the construction of its functional and structural cell components (Figure 3).
Transporter-coding genes: important components of the Ca. G. gigasporarum genome
Owing to its intracellular location, an efficient system of transporters that mediate the intimate interaction with the fungus is necessary for Ca. G. gigasporarum viability. Indeed, 150 CDS involved in transport functions were annotated.
A large set of proteins involved in phosphate, zinc and putrescine uptake were identified. The presence of these transporters suggests a strong dependency of Ca. G. gigasporarum on its AMF host, which, as for other Glomeromycota, is efficient in uptake of not only inorganic phosphate but also Zn (Gonzalez-Guerrero et al., 2005). Interestingly, a pathway for putrescine biosynthesis was demonstrated in the AMF Gigaspora rosea (Sannazzaro et al., 2004), suggesting that Ca. G. gigasporarum has the capability to take up this biogenic amine of fungal origin, and maybe to use it as a growth factor as suggested for other bacteria (Wortham et al., 2007).
As already discussed, evidence of amino-acid uptake by Ca. G. gigasporarum is given by the presence of many amino-acid transporters (Supplementary Text S3). A specific gene cluster coding for a dipeptide/heme/delta-aminolevulenic acid transporters (dpp operon) was identified. The dppA gene, the product of which is responsible for the specificity of the imported oligopeptides, is present in at least 20 copies, suggesting that peptide uptake is crucial for bacterial cell function. This hypothesis is strengthened by the annotation of a set of genes encoding proteases and peptidases targeted to the extracellular or periplasmic space, of which two secreted metallopeptidases share a strong similarity with sequences from the Gram-negative bacterium Stenotrophomonas maltophilia (Nyc and Matejkova, 2010). Ca. G. gigasporarum may release such peptidases to break down host proteins and then take up the resulting oligopeptides. Remarkably, electron microscopy studies have revealed that Ca. G. gigasporarum is often associated with fungal protein bodies (Bonfante et al., 1994): this peculiar location is consistent with Ca. G. gigasporarum exploiting fungal nitrogen resources via an efficient system of secreted peptidases and oligopeptide transporters.
Drug efflux is a general mechanism responsible for bacterial resistance to antibiotics: the Ca. G. gigasporarum genome contains at least nine genes involved in such a function, including an ABC-type antibiotic efflux pump and two gene clusters for the specific extrusion of macrolides (macAB) and acriflavine (acrAB). The presence of these genes most likely explains the failure to cure G. margarita from its endobacterium using antibiotics (Jargeat et al., 2004).
In summary, the presence of numerous transporter-coding genes suggests that Ca. G. gigasporarum has the mechanisms required to ensure mineral and nutritional supply from the fungal cell, where phosphorus and proteins are particularly abundant (Bonfante et al., 1994).
Interacting with the fungal host: secretion systems and virulence factors
Ca. G. gigasporarum proliferates inside a fungal phagosome/vacuole, suggesting that it communicates with the fungus via transport and secretion systems that deliver bacterial proteins to the host. This is supported by the large number of genes present in its genome coding for secretion systems. Among these, the complete Sec Protein secretion system machinery is present together with the related set of genes for the twin-arginine translocation pathway (Figure 3). Moreover, genes required for the type III secretion system (T3SS) were annotated and located in a genomic region that spans 17 771 bp, suggesting the organization in a pathogenicity island. T3SS is highly conserved in the genome of pathogenic and also symbiotic Gram-negative bacteria, where it acts as a molecular syringe that translocates effectors into the host cells. Phylogenetic analyses of five T3SS gene homologs have shown that the Ca. G. gigasporarum system has similarities to the SPI2 type (not shown): in most cases Ca. G. gigasporarum T3SS genes cluster with Chromobacterium violaceum ATCC 12472 and Yersinia pestis KIM homologs, which are closely related to the Salmonella genes located on SPI2 island (Deng et al., 2002). In contrast, BLASTP analysis shows that only a few T3SS genes of Ca. G. gigasporarum have similarity with homologous genes from the phylogenetically related endobacterium B. rhizoxinica, which instead possesses an hrp-type T3SS (Lackner et al., 2011b). Ca. G. gigasporarum also has genes that have been predicted to belong to T2SS and T4SS. In all, 8 out of 12 genes required for a functional type IV apparatus and involved in producing pili were annotated, in analogy to Hamiltonella defensa, which harbors a conjugative IncFII plasmid (Degnan et al., 2009). However, pili have never been observed in micrographs of Ca. G. gigasporarum when inside G. margarita; it is possible that Ca. G. gigasporarum only uses pili in specific stages of its life cycle.
Many of the T2SS, T3SS and T4SS genes of Ca. G. gigasporarum are expressed along the fungal life cycle (Supplementary Figure S4). Interestingly, the highest expression levels of gspD (general secretion pathway protein D) and secB (protein export chaperone SecB) transcripts were detected during the symbiotic phase (Supplementary Figure S4), which suggests a complex network of interactions among bacterial, fungal and plant phyla.
Conclusion
The work presented here is the first in-depth genome characterization of an endobacterium symbiont of a fungus, which itself is a symbiont of a plant. The 1.72-Mb genome of Ca. G. gigasporarum is strikingly reduced when compared with the free-living related Burkholderia species, whereas this feature is shared with many insect endosymbionts. The comparative genome analysis also revealed some functional similarities with these insect endobacteria, including the secondary facultative symbionts and those that are able to manipulate host reproductive functions (Akman et al., 2002; Foster et al., 2005; Moran et al., 2008). In addition, the prediction of the metabolic profile of Ca. G. gigasporarum unambiguously clusters it with insect endobacteria and not with a related fungal endosymbiont, Burkholderia rhizoxinica, which is a culturable microbe (Lackner et al., 2011a). These data suggest that Ca. G. gigasporarum has undergone a functional convergent evolution with phylogenetically distant endobacteria. We hypothesize that the driving force for such convergence is the strict nutritional dependence of Ca. G. gigasporarum on its fungal host, G. margarita.
Annotation of the Ca. G. gigasporarum genome revealed features that are typical of a symbiotic lifestyle (genome reduction, host-nutritional dependence). These traits are integrated with genetic determinants that are characteristic of other survival strategies, such as those of free-living bacteria, for example, pathways for vitamin B12 and antibiotic resistance. In addition, the presence of features like T3SS points to traits that were once considered pathogenic; however, increasing evidence shows that secretion systems may represent fitness factors also for symbionts (Deakin and Broughton, 2009).
Annotation of the Ca. G. gigasporarum genome provides an insight into the molecular basis for its obligate biotrophic status. The lack of some crucial metabolic pathways can explain the failure to grow Ca. G. gigasporarum as a free-living organism, as it has a metabolic dependence on the AMF host for both energy and nutrition. As the Ca. G. gigasporarum fungal host is itself an obligate biotroph that is dependent on its photosynthetic plant host, our work represents the first step towards uncovering the complex network of intimate interactions between plants, AMF and endobacteria. The data presented here provide clear evidence of the energy/nutrient flows of the tripartite interaction between the bacterium, the fungus and the plant (Figure 4). Ca. G. gigasporarum's limited capacity to synthesize amino acids, the presence of a large set of amino-acid transporters in the bacterial genome, and its location inside the protein-rich fungal vacuoles highlight that there is most likely a flow of nitrogen from the fungus to the bacterium. This view is well supported by the specific AMF capabilities to efficiently acquire nitrogen from organic matter (Hodge and Fitter, 2010) and to internalize it through specific transporters (Cappellazzo et al., 2008).

The drawing summarizes the most important nutritional and energetic fluxes that characterize the tripartite association of the endobacterium thriving inside the AM fungus that is inside a plant cell. For simplicity, the endobacterium is represented only once on the left side, although it is also present in fungal extraradical structures. The major fluxes are represented by N and P, which are taken up by the fungus and delivered to both the bacterium and the plant. On the other hand, organic carbon produced by the photosynthesis flows towards the fungus and then towards the endobacterium.
Analysis of the bacterial genome data reveals an entirely novel and intimate symbiosis between bacteria and symbiotic fungi, and identifies a previously unrecognized network of soil-dependent ecological interactions.
Accession codes
References
Akman L, Yamashita A, Watanabe H, Oshima K, Shiba T, Hattori M et al. (2002). Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat Genet 32: 402–407.
Anca IA, Lumini E, Ghignone S, Salvioli A, Bianciotto V, Bonfante P . (2009). The ftsZ gene of the endocellular bacterium ‘Candidatus Glomeribacter gigasporarum’ is preferentially expressed during the symbiotic phases of its host mycorrhizal fungus. Mol Plant Microbe Interact 22: 302–310.
Bianciotto V, Lumini E, Bonfante P, Vandamme P . (2003). Candidatus Glomeribacter gigasporarum. gen. nov., sp nov., an endosymbiont of arbuscular mycorrhizal fungi. Int J Syst Evol Microbiol 53: 121–124.
Bianciotto V, Genre A, Jargeat P, Lumini E, Becard G, Bonfante P . (2004). Vertical transmission of endobacteria in the arbuscular mycorrhizal fungus Gigaspora margarita through generation of vegetative spores. Appl Environ Microbiol 70: 3600–3608.
Bocs S, Cruveiller S, Vallenet D, Nuel G, Medigue C . (2003). AMIGene: Annotation of MIcrobial Genes. Nucleic Acids Res 31: 3723–3726.
Bonfante P, Anca IA . (2009). Plants, mycorrhizal fungi, and bacteria: a network of interactions. Annu Rev Microbiol 63: 363–383.
Bonfante P, Balestrini R, Mendgen K . (1994). Storage and secretion processes in the spore of Gigaspora margarita Becker and Hall as revealed by high-pressure freezing and freeze-substitution. New Phytol 128: 93–101.
Bontemps C, Elliott GN, Simon MF, Dos Reis FBD, Gross E, Lawton RC et al. (2010). Burkholderia species are ancient symbionts of legumes. Mol Ecol 19: 44–52.
Cappellazzo G, Lanfranco L, Fitz M, Wipf D, Bonfante P . (2008). Characterization of an amino acid permease from the endomycorrhizal fungus Glomus mosseae. Plant Physiol 147: 429–437.
Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F et al. (2010). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 38: D473–D479.
Castillo DM, Pawlowska TE . (2010). Molecular evolution in bacterial endosymbionts of fungi. Mol Biol Evol 27: 622–636.
Croft MT, Lawrence AD, Raux-Deery E, Warren MJ, Smith AG . (2005). Algae acquire vitamin B-12 through a symbiotic relationship with bacteria. Nature 438: 90–93.
Deakin WJ, Broughton WJ . (2009). Symbiotic use of pathogenic strategies: rhizobial protein secretion systems. Nat Rev Microbiol 7: 312–320.
Degnan PH, Yu Y, Sisneros N, Wing RA, Moran NA . (2009). Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc Natl Acad Sci USA 106: 9063–9068.
Deng W, Burland V, Plunkett G, Boutin A, Mayhew GF, Liss P et al. (2002). Genome sequence of Yersinia pestis KIM. J Bacteriol 184: 4601–4611.
Fitter AH, Helgason T, Hodge A . (2011). Nutritional exchanges in the arbuscular mycorrhizal symbiosis: implications for sustainable agriculture. Fungal Biol Rev 25: 68–72.
Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N et al. (2005). The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol 3: 599–614.
Gonzalez-Guerrero M, Azcon-Aguilar C, Mooney M, Valderas A, MacDiarmid CW, Eide DJ et al. (2005). Characterization of a Glomus intraradices gene encoding a putative Zn transporter of the cation diffusion facilitator family. Fungal Genet Biol 42: 130–140.
Hodge A, Fitter AH . (2010). Substantial nitrogen acquisition by arbuscular mycorrhizal fungi from organic material has implications for N cycling. Proc Natl Acad Sci USA 107: 13754–13759.
Hoffman MT, Arnold AE . (2010). Diverse bacteria inhabit living hyphae of phylogenetically diverse fungal endophytes. Appl Environ Microbiol 76: 4063–4075.
Jargeat P, Cosseau C, Ola’h B, Jauneau A, Bonfante P, Batut J et al. (2004). Isolation, free-living capacities, and genome structure of ‘Candidatus glomeribacter gigasporarum,’ the endocellular bacterium of the mycorrhizal fungus Gigaspora margarita. J Bacteriol 186: 6876–6884.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M . (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res 32: D277–D280.
Kuo CH, Ochman H . (2010). The extinction dynamics of bacterial pseudogenes. PLoS Genet 6: e1001050. doi:10.1371/journal.pgen.1001050.
Lackner G, Moebius N, Partida-Martinez L, Hertweck C . (2011a). Complete genome sequence of Burkholderia rhizoxinica, an endosymbiont of Rhizopus microsporus. J Bacteriol 193: 783–784.
Lackner G, Moebius N, Hertweck C . (2011b). Endofungal bacterium controls its host by an hrp type III secretion system. ISME J 5: 252–261.
Lumini E, Ghignone S, Bianciotto V, Bonfante P . (2006). Endobacteria or bacterial endosymbionts? To be or not to be. New Phytol 170: 205–208.
Lumini E, Bianciotto V, Jargeat P, Novero M, Salvioli A, Faccio A et al. (2007). Presymbiotic growth and sporal morphology are affected in the arbuscular mycorrhizal fungus Gigaspora margarita cured of its endobacteria. Cell Microbiol 9: 1716–1729.
McCutcheon JP, McDonald BR, Moran NA . (2009). Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc Natl Acad Sci USA 106: 15394–15399.
McCutcheon JP, Moran NA . (2010). Functional convergence in reduced genomes of bacterial symbionts spanning 200 my of evolution. Genome Biol Evol 2: 708–718.
Moller EM, Bahnweg G, Sandermann H, Geiger HH . (1992). A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res 20: 6115–6116.
Moran NA, McCutcheon JP, Nakabachi A . (2008). Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42: 165–190.
Naumann M, Schusler A, Bonfante P . (2010). The obligate endobacteria of arbuscular mycorrhizal fungi are ancient heritable components related to the Mollicutes. ISME J 4: 862–871.
Nyc O, Matejkova J . (2010). Stenotrophomonas maltophilia: significant contemporary hospital pathogen - review. Folia Microbiol (Praha) 55: 286–294.
Pereyre S, Sirand-Pugnet P, Beven L, Charron A, Renaudin H, Barre A et al. (2009). Life on arginine for Mycoplasma hominis: clues from its minimal genome and comparison with other human urogenital Mycoplasmas. PLoS Genet 5: e1000677.
Ruiz-Lozano JM, Bonfante P . (2000). A Burkholderia strain living inside the arbuscular mycorrhizal fungus Gigaspora margarita possesses the vacB gene, which is involved in host cell colonization by bacteria. Microb Ecol 39: 137–144.
Salvioli A, Lumini E, Anca IA, Bianciotto V, Bonfante P . (2008). Simultaneous detection and quantification of the unculturable microbe Candidatus Glomeribacter gigasporarum inside its fungal host Gigaspora margarita. New Phytol 180: 248–257.
Salvioli A, Chiapello M, Fontaine J, Hadj-Sahraoui AL, Grandmougin-Ferjani A, Lanfranco L et al. (2010). Endobacteria affect the metabolic profile of their host Gigaspora margarita, an arbuscular mycorrhizal fungus. Env Microbiol 12: 2083–2095.
Sannazzaro AI, Alvarez CL, Menendez AB, Pieckenstain FL, Alberto EO, Ruiz OA . (2004). Ornithine and arginine decarboxylase activities and effect of some polyamine biosynthesis inhibitors on Gigaspora rosea germinating spores. FEMS Microbiol Lett 230: 115–121.
Schmitz-Esser S, Tischler P, Arnold R, Montanaro J, Wagner M, Rattei T et al. (2010). The genome of the amoeba symbiont ‘Candidatus Amoebophilus asiaticus’ reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J Bacteriol 192: 1045–1057.
Smith SE, Read DJ . (2008). Mycorrhizal Symbiosis. Elsevier: London.
Taga ME, Walker GC . (2010). Sinorhizobium meliloti requires a cobalamin-dependent ribonucleotide reductase for symbiosis with its plant host. Mol Plant Microbe Interact 23: 1643–1654.
Tian CJ, Kasiborski B, Koul R, Lammers PJ, Bucking H, Shachar-Hill Y . (2010). Regulation of the nitrogen transfer pathway in the arbuscular mycorrhizal symbiosis: gene characterization and the coordination of expression with nitrogen flux. Plant Physiol 153: 1175–1187.
Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A et al. (2009). MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford). 2009:bap021; e-pub ahead of print 2009.
Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S et al. (2006). MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res 34: 53–65.
Wortham BW, Patel CN, Oliveira MA . (2007). Polyamines in bacteria: pleiotropic effects yet specific mechanisms. Adv Exp Med Biol 603: 106–115.
Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC et al. (2004). Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol 2: 327–341.
Zientz E, Dandekar T, Gross R . (2004). Metabolic interdependence of obligate intracellular bacteria and their insect hosts. Microbiol Mol Biol Rev 68: 745–770.
Acknowledgements
We thank Professor P Lammers (New Mexico State University) for his help in constructing the fosmid library, Dr E Oger for her help in annotation, Professor T Pawlowska (Cornell University) and Dr J Williams (Parco Tecnologico Padano) for the critical reading of the manuscript. This research was sponsored by the Converging Technologies project funded by the Regione Piemonte (CIPE-BIOBIT), and by the Compagnia di San Paolo to PB. SG was funded by Compagnia di San Paolo.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Ghignone, S., Salvioli, A., Anca, I. et al. The genome of the obligate endobacterium of an AM fungus reveals an interphylum network of nutritional interactions. ISME J 6, 136–145 (2012). https://doi.org/10.1038/ismej.2011.110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.110
Keywords
This article is cited by
-
Disentangling arbuscular mycorrhizal fungi and bacteria at the soil-root interface
Mycorrhiza (2023)
-
Upregulation of a marine fungal biosynthetic gene cluster by an endobacterial symbiont
Communications Biology (2020)
-
Narnaviruses: novel players in fungal–bacterial symbioses
The ISME Journal (2020)

{kind=link}