
Abstract
Tooth agenesis is described as the absence of one or more teeth. It is caused by a failure in tooth development and is one of the most common human developmental anomalies. We herein report genomic analyses of selective mandibular incisor agenesis (SMIA) using exome sequencing. Two Japanese families with SMIA were subjected to exome sequencing, and family with sequence similarity 65 member A (FAM65), nuclear factor of activated T-cells 3 (NFATC3) and cadherin-related 23 gene (CDH23) were detected. In the follow-up study, 51 Japanese and 32 Korean sporadic patients with SMIA were subjected to exome analyses, and 18 reported variants in PAX9, AXIN2, EDA, EDAR, WNT10A, BMP2 and GREM2 and 27 variants of FAM65, NFATC3 and CDH23 were found in 38 patients. Our comprehensive genetic study of SMIA will pave the way for a full understanding of the genetic etiology of SMIA and provide targets for treatment.
Similar content being viewed by others
Introduction
Humans usually develop 32 permanent teeth, including third molars. Human teeth are spatially specialized and classified as incisors and canines (the anterior teeth) and premolars and molars (the posterior teeth). Tooth agenesis denotes a pathological condition involving the absence of teeth because of a developmental failure. Selective (nonsyndromic) tooth agenesis is one of the most common dental anomalies; it also accompanies several syndromes, such as van der Woude syndrome, ectodermal dysplasias, oral–facial–digital syndrome type I, Rieger syndrome, holoprosencephaly and cleft lip and palate.1
Tooth agenesis most often occurs at the third molars across all populations.2 In populations of European descent, the second most common congenital missing teeth are the mandibular second premolars, followed by either the permanent maxillary lateral incisors3 or the maxillary second premolars.4,5 In Japanese patients and other Asians, the second most common missing teeth are the mandibular second premolars, followed by the mandibular and maxillary lateral incisors, and the maxillary second premolars.6–8 A notable difference has been observed in the frequencies of mandibular incisor agenesis between Japanese (1–2%)7,8 and European populations (0.2%).9
Selective tooth agenesis is known to be associated with variants of msh homeobox 1 (MSX1), paired box 9 (PAX9), axin 2 (AXIN2), ectodysplasin A (EDA), ectodysplasin A receptor (EDAR), EDAR-associated death domain (EDARADD), wingless-type MMTV integration site family member 10A (WNT10A),10 bone morphogenetic protein 2 (BMP2),11 gremlin 2 and DAN family BMP antagonist (GREM2).12 The identification of causality for agenesis of the mandibular incisors, referred to herein as selective mandibular incisor agenesis (SMIA), may enable us to elucidate the precise molecular mechanisms of temporal and spatial gene regulation in the distinctive development of human dentition. Therefore, in the current study, we performed exome analyses on 2 families and 83 sporadic patients of Japanese and Koreans demonstrating possible involvement of family with sequence similarity 65 member A (FAM65), nuclear factor of activated T-cells 3 (NFATC3) or cadherin-related 23 gene (CDH23) in the pathogenesis of SMIA in addition to the reported genes.
Materials and methods
Subjects
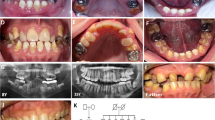
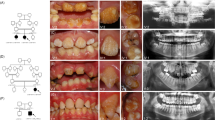
Patients with SMIA were diagnosed and recruited from the Showa University Dental Hospital (Tokyo, Japan) and affiliated hospitals. Standardized assessments, including panoramic radiographs, dental casts, intraoral photographs and anamnestic data, were performed in all patients. Dental specialists diagnosed the dentition status of all subjects and their available siblings and parents. Patients with developmental anomalies, such as ectodermal dysplasia, cleft lip or palate, or Down’s syndrome or those who had undergone orthodontic treatment previously were excluded from the study. This procedure has been considered reliable for diagnosing anomalies in tooth number in several studies.13,14
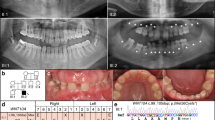
We recruited two Japanese family (Family A containing two affected and three unaffected individuals; Family B containing three affected and one unaffected individuals) showing a dominant transmission of SMIA (Figure 1). We also recruited 51 Japanese and 32 Korean patients with sporadic SMIA (aged 10–37 years). Twelve patients (SH3, SH14, SH23, SH24, SH31, SH35, SH38, SH49, SH50, SH51, SH66 and SH69) had agenesis of 2 incisors and 71 patients had agenesis of 1 incisor. The possibility of the misdiagnosis of agenesis of the mandibular central and lateral incisors caused by anatomical artifacts derived from the superposition of cervical vertebrae on the mental region should be noted.8 Therefore, in the present study, we did not distinguish agenesis between mandibular central and lateral incisors. Family histories of these patients were incomplete. Although the hearing ability of the patients with SMIA was not examined by an otolaryngologist, the dental doctor in charge confirmed that the patients could hold a normal conversation.

(a) Left; Family A, right; Family B. The pedigree showing autosomal-dominant inheritance of selective mandibular incisor agenesis. (b) Intraoral photographs and (c) panoramic radiograph of the proband (II:1) at 17 years of age. Note the absence of two mandibular incisors.
We collected saliva specimens from each patient and extracted DNA using the Oragene DNA Kit (DNA Genotek, Ottawa, Canada). This study was conducted under the approval of ethical committees at Showa University, Pusan National University and the National Institute of Genetics and performed according to the ethical principles defined in the Declaration of Helsinki. All subjects gave their informed consent to participate in the study.
Exome sequencing
Exome sequencing was performed for 5 individuals in Family A, 4 individuals in Family B and 83 sporadic patients (51 Japanese and 32 Koreans). DNA samples (3 μg) were subjected to exome capture using the SureSelect Human All Exon Kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s instructions. In brief, genomic DNA was randomly fragmented by sonication under standard conditions (Covaris, Woburn, MA, USA), followed by end repair, the addition of a single A base, adaptor ligation and gel electrophoresis to isolate 300-bp fragments, followed by PCR amplification. The captured DNA underwent high-throughput sequencing using the HiSeq2500 system (Illumina, San Diego, CA, USA).
Next, the size-selected libraries were used for cluster generation on the flow cells. All prepared flow cells were run on the Illumina HiSeq2500 using paired-end 100-bp reads. Reads were mapped to the reference genome (UCSC hg19) using Burrows-Wheeler Aligner v.0.7.9.15 Burrows-Wheeler Aligner-generated SAM files were converted to BAM format, then sorted and indexed using SAMtools v.0.1.18.16 Duplicated reads were marked with Picard v.1.102 (https://github.com/broadinstitute/picard). The files obtained in BAM format were analyzed using GATK v.2.7 following their best practice guidelines.17 In brief, BAM files were first subjected to insertion or deletion (indel) realignment, base quality score recalibration and variant calling with the UnifiedGenotyper walker to obtain potential variants in the Variant Call Format file. These variants were annotated using the algorithm (table.annovar.pl) in ANNOVAR (version 2013jul21).18 For gene annotation, we used the RefSeq gene database (build hg19),19 while variant annotation was based on dbSNP (dbSNP 137), the 1000 Genomes Project database20 and 1208 Japanese data in the Human Genetic Variation database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/index.html).
Filtering to detect causal variants
In Family A and Family B, variants detected from exome sequencing data were further analyzed by performing three filtering steps based on different criteria. In the first filtering step, we selected missense and nonsense variants, splice-site single-nucleotide variants and coding indels. The second filtering step was based on the frequency in the Human Genetic Variation database. Variants with a frequency <5% in the Human Genetic Variation database were filtered as SMIA candidates. Finally, heterozygous variants co-segregated in the family were selected. After these filtering steps, candidate variants were confirmed for all family members by Sanger sequencing on the 3730xl DNA Analyzer (Life Technologies, Carlsbad, CA, USA). Functional estimation and the conservation score of the variants were evaluated by prediction tools Polymorphism Phenotyping v221 and Genomic Evolutionary Rate Profiling,22 respectively.
In the 83 sporadic Japanese and Korean patients (SH1-SH83), variants of PAX9, AXIN2, EDA, EDAR, WNT10A, BMP2 and GREM2 previously reported in selective tooth agenesis were identified using exome data. Functional estimation and the conservation score of the variants were evaluated as before by Polymorphism Phenotyping v2 and Genomic Evolutionary Rate Profiling.
Results
Variants detection in families with exome sequencing
Exome sequencing was performed on five individuals in Family A, and on four individuals in Family B (Figure 1). The average coverage depth was 256.1×, with 98.8% of target bases covered by at least five reads. This supports a high level of confidence in the variant calling. As a result of the filtering procedure, three rare missense single-nucleotide variants in family with sequence similarity 65 member A (FAM65A), nuclear factor of activated T-cells 3 (NFATC3) and cadherin-related 23 (CDH23) were shown to be co-segregated in Family A and Family B in an autosomal-dominant manner.
Variant in the 83 sporadic patients
In 83 Japanese and Korean sporadic patients of SMIA, 33 patients had variants in the reported causal gene (Table 1). PAX9 variants (c.398A>G (n=1)), AXIN2 variants (c.1250C>T (n=4), c.1807G>C (n=6), c.1853A>G (n=1) and c.2213C>T (n=2)), EDA variants (c.428G>C (n=1), c.436G>T (n=1) and c.555T>A (n=1)), EDAR variants (c.319A>G (n=10), c.701G>T (n=1), c.1138A>C (n=2), c.1166G>C (n=1), c.1270G>A (n=1)), WNT10A variants (c.511C>T (n=1), c.637G>A (n=4), c.874A>G (n=3), c.909C>A (n=1)) and BMP2 variant (c.389G>A (n=1)). No variant was identified in MSX1, EDARADD or GREM2.
In 83 Japanese and Korean sporadic patients of SMIA, 38 patients have variants in FAM65, NFATC3 and CDH23 (Table 2). FAM65A variants (c.137C>T (n=5), c.148C>T (n=2), c.1117C>A (n=1), c.1994C>T (n=1), c.2542G>A (n=1), c.2668C>T (n=4), c.2734C>A (n=1) and c.2771G>A (n=5)), NFATC3 variants (c.244A>G (n=4), c.281A>C (n=1), c.1043C>T (n=1) and c.1279C>G (n=1)) and CDH23 variants (c.127G>A (n=9), c.141T>G (n=1), c.1637G>C (n=1), c.1681T>G (n=2), c.3179G>A (n=1), c.3352G>A (n=2), c.3397G>A (n=1), c.3656G>A (n=1), c.4009G>A (n=1), c.4135C>T (n=1), c.4346G>A (n=1), c.4762C>T (n=2), c.5722G>A (n=1) and c.6023G>T (n=1)).
Table 3 presented variants of candidate genes and scores of pathogenicity prediction tools.
Discussion
In the present study, we attempted to identify genetic causalities of SMIA, which is prevalent in Asian populations. Two family with SMIA was extensively analyzed by exome sequencing and co-segregating genes were screened in 83 sporadic patients of Japanese and Koreans. Exome analyses identified variants in the reported genes for tooth agenesis, including PAX9, AXIN2, EDA, EDAR, WNT10A and BMP2. In addition, newly identified genes, including FAM65, NFATC3 and CDH23, are reported as a possible causality.
Cadherin-23 is an atypical cadherin implicated in several deafness syndromes, including Usher syndrome, because of its role as a component of tip link structural proteins connecting sensory hairs of hair cells in the inner ear. CDH23 variants are thought to impair the structural maintenance of the tip link and to reduce contact between sensory hairs, resulting in nonsyndromic hearing loss.23 The functional role of FAM65A has been unknown. The product of NFATC3 is a member of the nuclear factors of activated T cells DNA-binding transcription complex. It is known to have a role in the regulation of gene expression in T cells and immature thymocytes.24
Experimental approaches in humans to identify genes that act on tooth development are limited. Therefore, the identification of genetic factors involved in defects of dentition such as tooth agenesis provides valuable information. Previous studies have shown that variants of MSX1 phenotypically lead to tooth agenesis mostly at the third molars, second premolars and maxillary first premolars, while PAX9 variants cause nonsyndromic tooth agenesis that preferentially affects the third molars, maxillary first and second molars and the mandibular second molars.25 Thus posterior teeth appear to be particularly sensitive to defects in MSX1 and PAX9.10 Conversely, autosomal-dominant variants of AXIN2 were shown to cause a severe form of tooth agenesis that preferentially affects the permanent molars, lower incisors and upper lateral incisors,26 while variants of EDA can lead to both hypohidrotic ectodermal dysplasia and nonsyndromic tooth agenesis favoring the anterior dentition.27 Thus the differential sensitivities of specific dentition types might depend on the differential expression of related genes and possibly reflect different roles for such genes during normal tooth development.10,25
Mouse teeth differ greatly from those of humans.28 For example, the labial side of the mouse incisor is coated with enamel and resembles a tooth crown, while the lingual side is more similar to a tooth root.29 Despite these differences in dentition, early stages of tooth development in both species are very similar, and the basis of tooth development and its molecular mechanisms originally discovered in the mouse have since been confirmed in humans.23,25,27 Knowledge from animal models, especially the mouse, is therefore important for our understanding of the genetic basis of tooth agenesis.30 Thus observation of Cdh23 mRNA expression in mouse mandibular incisors is also expected to occur in humans (Supplementary Figures S1A,B and S2I). We showed that Cdh23 is expressed in the central parts of the tooth buds and enamel organs and in the oral epithelia adjacent to the tooth germs only during the late bell stage (E16.5); it is not expressed in the maxillary incisors or mandibular molars in any stages (Supplementary Figure S2A–H). Bmp2 is expressed in the lower incisors only during the bell stage, although it is also expressed in the lower molars throughout the initiation stage, bud stage, cap stage and bell stage.31 Odontoblasts differentiate from the dental papilla and produce the dentin matrix during the bell stage, and ameloblasts simultaneously arise from the epithelium and secrete the enamel matrix.32 BMP2 reportedly has single-nucleotide polymorphisms that induce an increased risk of mandibular incisor agenesis.11
In summary, we found that novel variants in previously reported causal genes appear to mainly contribute to tooth agenesis of the anterior region, and the research using exome sequencing suggested the association between FAM65A, NFATC3 and CDH23 or SMIA. The early molecular diagnosis of tooth agenesis may enable to improve patient care29 and to alert clinicians to counsel patients to have regular colonoscopies in early diagnosis of AXIN2 mutations.30 Moreover, the identification of causality for agenesis of the mandibular incisors, referred to herein as SMIA, may enable us to elucidate the precise molecular mechanisms of spatial gene regulation in the distinctive development of human dentition. In this study, some patients exhibited a genetic causality that could not be determined. The expression of >200 genes within teeth were studied, and a large number of candidates were given.33 A limitation of this study was the relatively small number of pedigrees; thus replication with a larger-scale study could narrow down the number of these candidate genes. Further studies are also required to determine the contribution of FAM65A, NFATC3 and CDH23 to the causality of SMIA in different populations and the agenesis of human teeth other than the mandibular incisors.
References
Klein OD, Oberoi S, Huysseune A, Hovorakova M, Peterka M, Peterkova R . Developmental disorders of the dentition: an update. Am J Med Genet C Semin Med Genet 2013; 163: 318–332.
Rozkovcová E, Marková M, Dolejsí J . Studies on agenesis of third molars amongst populations of different origin. Sb Lek 1999; 100: 71–84.
Buenviaje TM, Rapp R . Dental anomalies in children: a clinical and radiographic survey. ASDC J Dent Child 1984; 51: 42–46.
Hunstadbraten K . Hypodontia in the permanent dentition. ASDC J Dent Child 1973; 40: 115–117.
Aasheim B, Ogaard B . Hypodontia in 9-year-old Norwegians related to need of orthodontic treatment. Scand J Dent Res 1993; 101: 257–260.
Davis PJ . Hypodontia and hyperdontia of permanent teeth in Hong Kong schoolchildren. Community Dent Oral Epidemiol 1987; 15: 218–220.
Endo T, Ozoe R, Kubota M, Akiyama M, Shimooka S . A survey of hypodontia in Japanese orthodontic patients. Am J Orthod Dentofacial Orthop 2006; 129: 29–35.
Goya HA, Tanaka S, Maeda T, Akimoto Y . An orthopantomographic study of hypodontia in permanent teeth of Japanese pediatric patients. J Oral Sci 2008; 50: 143–150.
Neal JJ, Bowden DE . The diagnostic value of panoramic radiographs in children aged nine to ten years. Br J Orthod 1988; 15: 193–197.
Arte S, Parmanen S, Pirinen S, Alaluusua S, Nieminen P . Candidate gene analysis of tooth agenesis identifies novel mutations in six genes and suggests significant role for WNT and EDA signaling and allele combinations. PLoS ONE 2013; 22: e73705.
Lu Y, Qian Y, Zhang J, Gong M, Wang Y, Gu N et al. Genetic variants of BMP2 and their association with the risk of non-syndromic tooth agenesis. PLoS ONE 2016; 11: e0158273.
Kantaputra PN, Kaewgahya M, Hatsadaloi A, Vogel P, Kawasaki K, Ohazama A et al. GREMLIN 2 mutations and dental anomalies. J Dent Res 2015; 94: 1646–1652.
Abe R, Endo T, Shimooka S . Maxillary first molar agenesis and other dental anomalies. Angle Orthod 2010; 80: 1002–1009.
Altug-Atac AT, Erdem D . Prevalence and distribution of dental anomalies in orthodontic patients. Am J Orthod Dentofacial Orthop 2007; 131: 510–514.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 2014; 42: D756–D763.
1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM et al. An integrated map of genetic variation from 1092 human genomes. Nature 2012; 491: 56–65.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Cooper GM, Stone EA, Asimenos G . NISC Comparative Sequencing Program, Green ED, Batzoglou S et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005; 15: 901–913.
Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG et al. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature 2004; 428: 950–955.
Stasik I, Rapak A, Zioło E, Strzadała L . The mitochondrial localization of RelB and NFATx in immature T cells. Cell Mol Biol Lett 2008; 13: 493–501.
Nieminen P . Genetic basis of tooth agenesis. J Exp Zool B Mol Dev Evol 2009; 312B: 320–342.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 2004; 74: 1043–1050.
Mues G, Tardivel A, Willen L, Kapadia H, Seaman R, Frazier-Bowers S et al. Functional analysis of Ectodysplasin-A mutations causing selective tooth agenesis. Eur J Hum Genet 2010; 18: 19–25.
Miletich I, Sharpe PT . Normal and abnormal dental development. Hum Mol Genet 2003; 12: R69–R73.
Thesleff I, Tummers M . Tooth organogenesis and regeneration. In: StemBook. The Stem Cell Research Community: Cambridge, MA, USA, 2009. http://www.stembook.org.
Prasad MK, Geoffroy V, Vicaire S, Jost B, Dumas M, Le Gras S et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J Med Genet 2016; 53: 98–110.
Gene expression in tooth maintained by Tooth and Craniofacial Development Group of the Developmental Biology Programme, Institute of Biotechnology, University of Helsinki. http://bite-it.helsinki.fi/.
Thesleff I, Hurmerinta K . Tissue interactions in tooth development. Differentiation 1981; 18: 75–88.
Nieminen P, Pekkanen M, Aberg T, Thesleff I . A graphical WWW-database on gene expression in tooth. Eur J Oral Sci 1998; 106(Suppl 1): 7–11.
Acknowledgements
We thank the participants of this study. This work was partly supported by a KAKENHI Grant-in-Aid for challenging Exploratory Research 25670881 (to TY), a KAKENHI Grant-in-Aid for challenging Exploratory Research 25670880 (to KM), a KAKENHI Grant-in-Aid for Scientific Research (A) 25251042 (HI), a Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT)-Supported Program for the Strategic Research Foundation at Private Universities, 2012–2017 and a Grant-in-Aid for Scientific Research on Innovative Areas ‘Genome Science’ from MEXT (No 221S0002).
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yamaguchi, T., Hosomichi, K., Yano, K. et al. Comprehensive genetic exploration of selective tooth agenesis of mandibular incisors by exome sequencing. Hum Genome Var 4, 17005 (2017). https://doi.org/10.1038/hgv.2017.5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.5
This article is cited by
-
EDA ligand triggers plasma membrane trafficking of its receptor EDAR via PKA activation and SNAP23-containing complexes
Cell & Bioscience (2023)
-
Characterisation of a second gain of function EDAR variant, encoding EDAR380R, in East Asia
European Journal of Human Genetics (2020)
