
Abstract
Familial Mediterranean fever (FMF) is a hereditary autoinflammatory disease that is inherited in an autosomal recessive manner and is caused by mutations in the MEFV gene. As the name indicates, FMF occurs within families and is more common in individuals of Mediterranean descent than in persons of any other ethnicity. To date, 314 mutations have been reported. We studied a Moroccan family with a total of five members, including a mother who was presenting with symptoms of FMF, while her four children remained asymptomatic. The five patients were screened by DNA sequencing of exon 2 and exon 10 of the MEFV gene. Then, complete exome sequencing analysis of the MEFV gene was done for the patients in whom a novel mutation was detected. This analysis identified a novel single base Cytosine (C) insertion mutation in the coding region of the MEFV gene, named c.441dupC (p. Glu148Argfs*5 or E148RfsX5), which resulted in a mutated Pyrin/Marenostrin protein. This is the first report of a new mutation in exon 2 of the MEFV gene in a Moroccan family. This novel insertion mutation may provide important information for further studies of FMF pathogenesis.
Similar content being viewed by others
Introduction
Familial Mediterranean fever (FMF), which belongs to a family of autoinflammatory diseases, remains a major health threat to the Mediterranean population, especially Sephardic Jews, North African Arabs, Armenians, Turks, Greeks, and Italians.
However, in recent years, an increasing number of cases have been reported in countries not near this area, such as the United States and Japan. Clinically, FMF is characterized by recurrent attacks of fever with pleuritis, skin lesions, abdominal pain, peritonitis, and amyloidosis (Amyloid A ‘SAA’).1,2
The symptoms and severity vary among affected individuals, sometimes even among members of the same family. FMF is usually inherited in an autosomal recessive manner1,3 and is caused by mutations of the MEFV gene on the short arm of chromosome 16 at position p13.3. MEFV comprises 10 exons and normally encodes a 781-amino acid protein, named pyrin or marenostrin.2,4 Gene mutations result in defective pyrin molecules; it is hypothesized that altered pyrin cannot suppress minor and unknown triggers to inflammation that are normally inhibited by intact pyrin. To date, more than 314 gene mutations and polymorphisms have been discovered in the MEFV gene.5
In this study, we examined five related patients. The mother of the family was homozygous for this mutation and was presenting with FMF symptoms. However, her four children were heterozygous for this mutation and did not show any clinical features of FMF.
Materials and methods
Subjects
Approximately 300 clinically pre-diagnosed FMF cases are referred to our laboratory for the detection of MEFV mutations each year. Among those patients with clinical evidence of FMF, five individuals from a Moroccan family were studied.
PCR amplification and sequencing
After obtaining informed consent, peripheral blood was collected from each patient and genomic DNA samples were extracted from blood lymphocytes using the Gene Cather Magnetic Beats kit (Invitrogen, Carlsbad, CA, USA). For each patient, both MEFV exon 2 and 10, which are considered as mutation hot spots,6 were individually amplified byPCR using 2 pairs of corresponding primers:
Exon 2: F: 5′- GCCTGAAGACTCCAGACCACCCCG-3′, R: 5′- AGGCCCTCCGAGGCCTTCTCTCTG-3′
Exon 10: F: 5′- GAGGTGGAGGTTGGAGACAA-3′, R: 5′- TGACCACCCACTGGACAGAT-3′. PCR was performed in a 25 ml reaction volume containing 60 ng of genomic DNA, 5 U of Taq (Invitrogen), 20 pmol of each primer, 50 mM MgCl2, 10 mM d NTP, and 10× PCR buffer (Invitrogen) in the Veriti 96-well Thermal Cycler 9902 (Applied Biosystems, Foster City, CA, USA). The PCR conditions were as follows: initial denaturation at 94 °C for 5 min, 35 cycles at 94 °C for 30 s and 58 °C for 45 s, 72 °C for 1 min, and a final extension at 72 °C for 5 min. Bidirectional direct sequencing of purified PCR products was performed using the BigDye Terminator V1.1 Cycle Sequencing kit (ABI prism, Foster City, CA, USA) and an Applied Biosystems 3500DX Genetic analyzer.
The resulting chromatogram was analyzed using the Sequencing Analysis SeqA V5.4 (Applied Biosystems) program. The obtained sequences underwent bioinformatics analysis using the ‘Nucleotide Blast’ Alignment Program at http://blast.ncbi.nlm.nih.gov/Blast.cgi.
Results
Our analysis of five related Moroccan patients revealed that the mother of this family (age 35 years) presented with the clinical features of FMF, including recurrent attacks of fever, abdominal pain, and skin lesions.
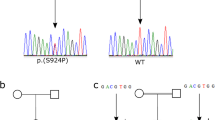
Indeed, the routine analysis of MEFV mutations in this family conducted in our laboratory clearly showed a single C nucleotide duplication, resulting in a frameshift mutation in exon 2 at the 148th codon (between the 441st and 442th nucleotides). The frameshift mutation was confirmed by DNA sequencing of the children as well as their parents, and the results indicated the existence of this mutation in the mother and her children (Figures 1a and c), but the absence of this mutation in the father (Figure 1b).

Electrophoregrams of the new homozygous and heterozygous insertion at the 441st nucleotide of MEFV as well as the wild type. The insertion resulted in a frameshift in codon 148. (a) Forward electrophoregram for the homozygote (Mother); (b) Forward electrophoregram for the wild type (Father); (c) Forward electrophoregram for the heterozygotes (Children).
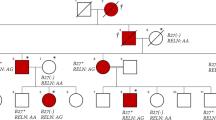
Our findings also showed that the four children of this Moroccan family were mutation c.441dupC (p. Glu148Argfs*5 or E148RfsX5) carriers. It is well documented that mutations of the MEFV gene cause FMF in an autosomal recessive manner; therefore, the heterozygous mutation carriers should not become symptomatic at any point in their lives (Table 1). The family tree of these five individuals and the detected mutations are illustrated in Figure 2. The C nucleotide insertion and frameshift mutation between the 441st and 442th nucleotides caused the conversion of the nucleotide sequence from 5′-CCCGAGGCC … CGACCTAGA-3′ to 5′-CCCCGAGGC … GCGACCTAG-3′ and this conversion resulted in a stop at codon 241.

Genealogical tree of the studied patients.
The protein sequence converted from Glu-Ala-Gly … Met-Arg-Pro-Arg to Arg-Gly-Arg … Asp-Ala-Thr-STOP (E148RfSX5 or Glu148Argfs*5). The c.441dupC mutation was searched for using six different web-based mutation analysis tools. MutationTaster, PolyPhen2 (HumDiv prediction version), and Ensembl.org indicated that the c.441dupC mutation might have pathogenic effects (Table 2).7,8,9,10
Children
A 15-year-old male (child (1) a 10-year-old female (child (2) an 8-year-old female (child (3) and a 3-year-old female (child (4) all without FMF symptoms; c.441dupC was detected by sequencing of the 4 children (Figure 1c).
Discussion
The main findings of this investigation have shown for the first time a novel frameshift mutation in the MEFV gene (c.441dupC) that was detected in 5 FMF patients from one Moroccan family. Analysis of the clinical and genetic features of the mother showed that she was homozygous for the c.441dupC mutation and presented with the clinical symptoms that are associated with FMF. In addition, the children were heterozygous for the c.441dupC mutation and were asymptomatic because, in autosomal recessive inheritance, two copies of a disease allele are required for an individual to be susceptible to expressing the phenotype. The four asymptomatic children had one copy of a disease allele and one copy of a normal allele and, as a result, they are carriers but do not show symptoms of the disease.
This novel mutation was detected in exon 2 of the MEFV gene, where major mutations that have been identified and described as being responsible for FMF are located. Moreover, each functional region of MEFV encodes part of the pyrin protein. Exon 1 encodes a domain named the pyrin domain (PYD). The bZIP is encoded by exon 2, an alpha helical region is encoded by exons 3–8, and a domain called B303/SPRY is encoded by exon 10.11
In addition, each domain of the pyrin protein is responsible for many protein-protein interactions: the bZIP domain, which is encoded by exon 2, interacts with p65 and Ikβ-α.12
The existence of a bZIP transcription factor basic domain and two nuclear localization signals, which are encoded by a basic residue cluster PLESKREF beginning at 150-amino acid residue and a bipartite NLS motif at residues 420–437, suggests that pyrin acts as a nuclear factor.13
However, in transfected cells, full-length pyrin exclusively localizes to the cytoplasm, while isoforms lacking exon 2 can enter the nucleus.14 Nevertheless, immunostaining of various pyrin-expressing cells showed that endogenous pyrin is predominately localized to the nucleus in synovial fibroblasts, dendritic cells, and polymorphonuclear cells, but to the cytoplasm in monocytes.15 To date, 101 mutations have been detected in exon 2, the most recent of which was identified in 2016 and was a deletion called C.382-390del (P. Glu-128-As 130del).5
The mother was homozygous for the mutation since she carried two copies of the mutated allele, while her children were heterozygous. This novel frameshift mutation is located in exon 2 of the MEFV gene and causes the replacement of amino acids after the 148th amino acid, resulting in a new amino acid sequence of Glu148Arg—Ala149Gly— ……… -Pro240Thr—Arg241STOP. Previously identified FMF causing mutations are also located in exon 2 of the MEFV gene. The insertion described in this article is significant because it is the only insertion and frameshift mutation reported so far in exon 2 of the MEFV gene.
Conclusions
Our study is the first to show a novel insertion in exon 2 of the MEFV gene through the examination of the clinical and genetic features of five related Moroccan patients who were clinically suspected of having FMF. Thus, we suggest that this novel insertion mutation could provide important information for further studies on FMF pathogenesis.
References
Touitou I, Sarkisian T, Medlej-Hashim M, Tunca M, Livneh A, Cattan D et al. International study group for phenotype-genotype correlation in familial mediterranean fever. country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum 2007; 56: 1706–1712.
Aldea A, Campistol JM, Arostegui JI, Rius J, Maso M, Vives J et al. A severe autosomal-dominant periodic inflammatory disorder with renal AA amyloidosis and colchicine resistance associated to the MEFV H478Y variant in a Spanish kindred: an unusual familial Mediterranean fever phenotype or another MEFV-associated periodic inflammatory disorder? Am J Med Genet A 2004; 124: 67–73.
The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90: 797–807.
The French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17: 25–31.
Infevers 2016. Available at: http://fmf.igh.cnrs.fr/ISSAID/infevers/. (accessed 10 September 2016)
Touitou I . The spectrum of familial Mediterranean fever (FMF) mutations. Europ. J Hum Genet 2001; 9: 473–483.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F . Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics 2010; 26: 2069–2070.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Kumar P, Henikoff S, Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081.
McLaren W, Gil L, Hunt S, Riat H, Ritchie G, Thormann A et al. The ensembl variant effect predictor. Genome Biol 2016; 17: 122.
Yepiskoposyan, Levon, and Harutyunyan, Ashot (eds) Molecular Genetics of Familial Mediterranean Fever. (John Wiley & Sons Ltd, Chichester, UK, 2008). Available at: http://www.els.net [doi: 10.1002/9780470015902.a0021442].
Chae JJ, Komarow HD . Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell 2003; 11: 591–604.
Centola M, Aksentijevich I, Kastner DL . The hereditary periodic fever syndromes: molecular analysis of a new family of inflammatory diseases. Hum Mol Genet 1998; 7: 1581–1588.
Papin S, Duquesnoy P, Cazeneuve C . Alternative splicing at the MEFV locus involved in familial Mediterranean fever regulates translocation of the marenostrin/pyrin protein to the nucleus. Hum Mol Genet 2000; 9: 3001–3009.
Diaz A, Hu C . Lipopolysaccharide induced expression of multiple alternatively spliced MEFV transcripts in human synovial fibroblasts: a prominent splice isoform lacks the C-terminal domain that is highly mutated in familial Mediterranean fever. Arthritis Rheum 2004; 50: 3679–3689.
Acknowledgements
We thank each member of the family for their generous participation in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Mejtoute, T., Sayel, H., El-Akhal, J. et al. The detection of a novel insertion mutation in exon 2 of the MEFV gene associated with familial mediterranean fever in a moroccan family. Hum Genome Var 4, 17023 (2017). https://doi.org/10.1038/hgv.2017.23
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.23
