
Abstract
In sheep, coat colour (and pattern) is one of the important traits of great biological, economic and social importance. However, the genetics of sheep coat colour has not yet been fully clarified. We conducted a genome-wide association study of sheep coat colours by genotyping 47 303 single-nucleotide polymorphisms (SNPs) in the Finnsheep population in Finland. We identified 35 SNPs associated with all the coat colours studied, which cover genomic regions encompassing three known pigmentation genes (TYRP1, ASIP and MITF) in sheep. Eighteen of these associations were confirmed in further tests between white versus non-white individuals, but none of the 35 associations were significant in the analysis of only non-white colours. Across the tests, the s66432.1 in ASIP showed significant association (P=4.2 × 10−11 for all the colours; P=2.3 × 10−11 for white versus non-white colours) with the variation in coat colours and strong linkage disequilibrium with other significant variants surrounding the ASIP gene. The signals detected around the ASIP gene were explained by differences in white versus non-white alleles. Further, a genome scan for selection for white coat pigmentation identified a strong and striking selection signal spanning ASIP. Our study identified the main candidate gene for the coat colour variation between white and non-white as ASIP, an autosomal gene that has been directly implicated in the pathway regulating melanogenesis. Together with ASIP, the two other newly identified genes (TYRP1 and MITF) in the Finnsheep, bordering associated SNPs, represent a new resource for enriching sheep coat-colour genetics and breeding.
Similar content being viewed by others

Introduction
The physical basis of variation in coat colour (and pattern) of domestic sheep (Ovis aries) is the ratio and amount of melanin (either eumelanin or pheomelanin) pigments produced in the cells of the wool follicles and transported into hair, skin and wool (see the review by Cockett et al. (2001) and Deng et al. (2009)). Domestication has changed the morphology as well as genetics associated with variations in coat pigmentation in sheep (Kijas et al., 2012). The standard wild-type coat colour in sheep is generally a dark body with a pale belly (Sponenberg, 1997), which is relatively rare in domestic sheep. Coat colour is a key breed characteristic and economically important production trait in sheep, unlike in their wild mouflon (Ovis aries orientalis) ancestors where the lack of artificial selection for coat colour allows variants to arise and segregate (Norris and Whan, 2008). After a long-term strong artificial selection for white fibres, the phenotype of white coat colour has accumulated into a high frequency in some sheep breeds and shows a pattern of autosomal dominant inheritance (Adalsteinsson, 1970; Li et al., 2011). Sheep coat colour was considered to be a independent single locus trait and varies substantially within and across populations, from the lightest white (for example, the Royal White sheep in the United States) to the darkest brown (the Brown Mountain sheep in Switzerland) and black (for example, the Black Welsh Mountain sheep).
Earlier research on sheep colour genetics progressed slowly because most ewes produce 1–2 lambs a year and classic segregation analyses were therefore not effective or concordant (Davenport, 1905; Warwick et al., 1957; Adalsteinsson, 1970). Recently, application of newly developed molecular methods allowed new insights into sheep colour genetics and indicated variation at a number of main candidate genes on different chromosomes (for example, agouti signalling protein (ASIP) on O. aries chromosome 13 (OAR13); the candidate genes identified to date are detailed in Supplementary Table S1) implicated in sheep coat colour. The candidate genes are involved, for example, in regulating melanogenesis, coding for melanogenic enzymes, and mediating melanocyte development and migration during embryogenesis (Jackson, 1994; Slominski et al., 2004; Costin and Hearing, 2007).
The Finnsheep is an ancient, short-tailed breed, with ancestries in the most original type of sheep that first immigrated into and spread from the Near-Eastern domestication centre to Europe (Chessa et al., 2009). Long-term geographic and genetic isolation has left the population of Finnsheep in Finland with genetic characteristics such as a relatively limited effective population size (Ne=ca. 120; Li et al., 2009), which is less than that for most modern sheep breeds (Ne>300; Kijas et al., 2012). In general, the crossbreeding has been used in the Finnsheep, and, thus, currently <15 000 ewes and 1000 breeding rams are left in Finland (Li et al., 2009). Finnsheep have been farmed, regularly phenotyped (for example, the litter size and coat colour) and the pedigrees have been recorded. The Finnsheep population in Finland shared several features (for example, high level and wider intervals of linkage disequilibrium (LD)) in common with genetically isolated populations, making them a useful genetic resource for studies aimed at improved understanding of the genetic basis underlying complex traits, particularly for mapping recessive genes for the traits (see the reviews by Peltonen et al. (2000) and Kristiansson et al. (2008)). Strikingly, substantial substructure was detected among the Finnsheep of different colours based on microsatellite and genome-wide single-nucleotide polymorphism (SNP) analyses (for example, Tapio et al., 2003; Li et al., 2011), which may increase the chance of producing a false-negative result (type II error) in genome-wide association studies (GWAS). Nevertheless, this genetically heterogeneous population offers a unique opportunity to detect the genetic hitchhiking effect (or selection sweeps) of a favourable gene within a population (for example, Li and Merilä, 2010).
The Finnsheep breed is of multiple colours and patterns (‘coat colour pattern’ defines how the coat colour is distributed or concentrated on the body), which mainly include white (ca. 65%; see Figure 1a), grey (ca. 5%; the grey is the mixture of white and black hair; see Figure 1b), black (ca. 20%) and brown (ca. 9%; Puntila, 1997; Puntila et al., 1999; see Figure 1a). The coat colours and patterns (or spotting) in the Finnsheep are comparable with those in the Icelandic sheep, where three loci (the basic colour gene B, the pattern gene A and the spotting locus S) are involved in the dominant and recessively coloured coat phenotypes (for example, Adalsteinsson, 1970; Reinieri et al., 2008), while little has been understood about the molecular genetic cause of coat colour variation in the Finnsheep. In the meantime, the list of candidate genes associated with coat colour and skin pigmentation in other domestic animals and humans has been supplemented with additional inputs such as FGF5, TP53BP1, SLC24A4, OCA2 and HERC2 by using the GWAS (for example, Sulem et al., 2007; Han et al., 2008; Cadieu et al., 2009). These findings have enabled not only an increasingly detailed genetic dissection of the pigmentation and colour phenotypes but also the realization that the phenotypes could be much more complex than previously imagined and controlled by a gene network rather than single genes. Identifying the genetic determinants of coat colour in sheep is critical for improving the efficiency of artificial selection for the desired coat colour (or pattern) as well as for understanding different evolutionary dynamics (for example, directional selection and genetic drift) in shaping the phenotypic variation in farm and wild animals (see for example, Johnston et al., 2011; Gratten et al., 2012).

Coat colour and pattern polymorphism in the Finnsheep. Black (a, left), brown (a, middle), white (a, right), grey (b) and black/white spotted (c).
To date, all investigations for genes associated with sheep coat colours have used linkage (for example, Våge et al., 2003; Beraldi et al., 2006) and candidate gene analytic methods (for example, Loehr et al., 2008; Norris and Whan, 2008; Gratten et al., 2010). Whereas linkage analysis is a useful tool for identifying rare genetic variants with relatively large phenotypic effects that follow Mendelian laws of inheritance, GWAS is the method of choice for detecting common genetic variants associated with phenotypic variation (for example, Kayser et al., 2008), and a higher resolution can be achieved. We performed a GWAS in the Finnsheep in Finland, an isolated sheep population, using data on 47 303 SNPs created as part of the Sheep Hapmap project, coordinated by the International Sheep Genomics Consortium (ISGC; http://www.sheephapmap.org/hapmap.php). By performing a genome-wide selection test, we also wanted to characterize the patterns of genomic variation that artificial selection for coat colour has imparted on the genome of sheep.
Materials and methods
Animals and coat colour
We collected blood samples from 99 adult Finnsheep of different coat colours or patterns. The samples were from different flocks in order to cover as complete lineage of the Finnsheep as possible. Only the animals with detailed pedigree records and in typical coat colours of the Finnsheep have been sampled. Information on coat colour of the samples was documented and the samples were grouped into five categories of typical colours and patterns (white, grey, brown, black and black/white spotted; nwhite=54, ngrey=14, nbrown=16, nblack=14, nblack/white spotted=1; 45 males and 54 females; see Figure 1).
DNA extraction, genotyping and quality control
We extracted the sheep genomic DNA from the whole blood using standard methods with the Puregene Blood Kit (Qiagen Nordic, Helsinki, Finland). As coordinated by the ISGC, the ovine SNP50K BeadChip genotyping of the DNA samples were carried out via the Illumina technology (Illumina, San Diego, CA, USA). Details on creation of the ovine beadchip and genotyping procedures are available in the ovine SNP50 HapMap data set (http://www.sheephapmap.org/hapmap.php), Kijas et al. (2009) and Miller et al. (2011).
Markers were excluded based on assay abnormalities annotated by Illumina, segregation inconsistencies documented by ISGC or MAF=0. Additional standard quality control steps have been also performed. These included: exclusion of SNPs with high rates of missing alleles, with different missing genotype rates between sheep in different colours (ΔP=P1–P2>5%; P1 and P2 are the missing genotype rates of sheep in two different colours, respectively) or the significance value from a test of Hardy–Weinberg equilibrium <0.0001. We further excluded the SNP markers with either chromosome or base-pair position within a chromosome, or both unknown; thus, 47303 SNPs were kept in the subsequent analysis.
All individuals genotyped have the call rate >98% (that is, the fraction of the number of SNPs with a genotype call per sample over the total number of SNPs on the Beadchip) with an overall value (that is, the value of call rates averaged by the total number of individuals) of 99.99%. We examined the individuals which are in discrepancy between their reported sex and the sex inferred from the X chromosome, being indicative of sample mix-ups. Further, we identify individuals closely related at the level of half-sibs or closer (for example, parent–offspring or full-sibs) and sample duplications (Li et al., 2011) using the identity-by-descent option of the PLINK programme. No case was found in the examinations of discordant gender and sample duplication. Based on results of the relatedness analysis above, one individual from each of the closely related pairs was excluded in the analysis. In the case that a single individual was involved in the indicated relatedness with several other individuals, this individual was excluded. Finally, a set of 91 samples (nwhite=50, ngrey=13, nbrown=14, nblack=13, nblack/white spotted=1) was available for the analyses.
Assessment of population stratification
We used the genomic-control (GC) method (Devlin and Roeder, 1999), which was based on a Bayesian outlier model, to adjust for population stratification in the sample. In an earlier study (for example, Li et al., 2011), three subpopulations corresponding roughly to the different coat colours (grey, white and black and brown) were identified in the total of 99 Finnsheep based on the full panel of SNP markers (n=49 034). A global FST=5.4% (P<0.05) is diagnostic of differentiation between the three coat-colour subpopulations (for example, Li et al., 2011).
To account for the possibility of spurious associations caused by population stratification, the significant P-values of the following association analyses were corrected using the GC parameter, λGC (for example, Bacanu et al., 2002). Evidence of systematic biases (see Results; λGC=2.323) was indicated in the initial analyses without adjustment for any variables. For the 47 303 SNPs in the GWAS, the distribution of observed P-values with those expected under the global null hypothesis (that is, no association between the tested SNPs and coat colours) was compared using the Q–Q plot (quantile–quantile plot). The PLINK software (Purcell et al., 2007) was applied in the approach. The GC method of correcting for population stratification is generally used within single populations. The Finnsheep studied here is a single population, but given the strong and significant population sub-structuring within the Finnsheep population in this study, we implemented the analyses again with the EIGENSTRAT method (Price et al., 2006) and the adjusted top four principal components (PCs) of genetic variation as implemented in EIGENSOFT (Patterson et al., 2006). This gave similar results as that with the adjustments with the GC method.
Association, segregation model and LD analyses
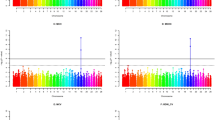
For the genome-wide association (GWA) analysis of coat colour we regressed an ordinal coding for the coat colours (white=1, grey=2, brown=3, black=4 and black/white spotted=5; for the different sheep coat colours, see Figure 1) on an ordinal coding for genotypes (0, 1 or 2 copies of the minor allele of a SNP) for SNPs individually using a linear model in the PLINK software (Purcell et al., 2007). The significant P-values for association from the linear regression analysis were corrected for known population structure by adjusting for the first four components. An empirical P-value less than the stringent level of 0.01 was set for the genome-wide significance. The threshold value (nominal P<2.11 × 10−7=0.01/47303; see the RESULTS) was determined by applying the Bonferroni correction accounting for multiple hypotheses testing. The Manhattan plot of −log10(observed P-value) from GWAS analysis was produced using the ggplot2 (Wickham, 2009) in the R package.
It was early evidenced that determinants of sheep coat colour may act along two phenotypic axes: white coat versus non-white coat colour. For example, alleles at the ASIP gene were found primarily to determine presence or absence of white coat colour (Norris and Whan, 2008). Hence, we implemented additional analyses among individuals without white coat colour and compared those with white coat coloured individuals to evaluate whether there were loci at one or both of the phenotypic axes. The ordinal coding for coat colour was regressed on counts of minor alleles after excluding white-coated individuals and logistic regression was used to test the association between the ordinal genotype coding and binary white-coated phenotypes (white versus non-white coat colour; refer to the method in the study by Han et al. (2008) for details). The analyses were conducted in a categorical way with white versus non-white.
In addition, we used the χ2 tests for goodness-of-fit to assess the dominant or recessive model of gene action as follows (Mather, 1963),

where z is the expected ratio of phenotypic segregation, the observed numbers of phenotypes are a1 and a2 and n is the total number of observations. If the χ2 value is <0.05, the data were not deemed to agree with the proposed segregation ratio (also refer to the study by Lambrides et al. (2004)). A cohort (Li et al., 2011) containing parents (P1, white, genotype AG; P2, white, genotype AG) and 12 F1 offspring (white=8 individuals (AA=3, AG=5); non-white=4 individuals (GG=4)) was included in the test.
We calculated the LD coefficients (r2) between individual significant SNPs with P<2.11 × 10−7 and estimated the frequencies of the four possible two-SNP haplotypes. Further, we used the four-gamete rule (for example, Wang et al., 2002) to identify haplotype blocks. In the four-gamete rule approach, a recombination event is deemed to have occurred if all the four haplotypes showed a frequency of ⩾0.01. Haplotype blocks were then constituted of consecutive SNPs where only three gametes were detected. LD metrics between SNPs were from the Sheep HapMap, displayed using Haploview (v4.2) and SNAP. LD blocks were defined using the same method for defining human haploblocks used in HapMap release 27 (also refer to the studies by Gabriel et al. (2002) and Myers et al. (2007)).
Detection of selection signals
To assess the effect of genetic selection for white (versus non-white) coat colour on the genome-wide SNPs, we used a robust Bayesian test (Beaumont and Balding, 2004) as implemented in the programme LOSITAN (http://popgen.eu/soft/lositan/). In the programme, we applied 100 000 simulated loci, giving an expected neutral distribution of FST values and an estimate at the 0.05 significance for each SNP. Each simulation included 50 individuals per population, 2 populations, 47 303 loci and an expected FST value of 5.92%. This method detected SNPs under selection by examining the genome-wide distributions of FST values higher (positive selection) or lower (balancing selection) than expected and the significance level (P-values; Beaumont and Balding, 2004). Both ‘neutral mean FST’ and ‘force mean FST’ options were employed as suggested by Antao et al. (2008). In the outlier selection tests, we did the tests between the two groups of individuals (white versus non-white). We further used a five-SNP sliding window to calculate the average significance P-values across each chromosome to detect specific genomic regions putatively under strong and recent selective sweeps. The average P-values for the consecutive five SNPs were coded at a critical level of 0.05, for example if the average P-values for consecutive five SNPs were <0.05, they were coded as categorical variable 1; otherwise they were coded as 0. Following this procedure, the SNP windows with significant P-values <0.05 were identified. Adjacent signatures were considered as ‘genomic regions’ if they included 10 consecutive windows with significant average P-values (P<0.05).
Bioinformatics
As the sheep genome annotation is not yet complete (Dalrymple, 2007; Becker et al., 2010), the gene annotation of the mapped interval was inferred from the corresponding human and bovine Reference Sequence (RefsSeq), both of which were converted from the UMD3 bovine genome assembly (http://www.bovinegenome.org). All the positions correspond to the Real Sheep Genomes Oar v3.1 (http://www.livestockgenomics.csiro.au/sheep/oar3.1.php). The corresponding human and bovine chromosomal segment was obtained by BLASTN searches of the ovine SNP flanking sequences on the human and bovine genome through the Real Sheep Genome Oar v3.1. In this analysis, we defined the ‘targeted gene region’ as being ∼500 kb around the significant SNP (significant SNP ±500 kb; for example, Scherag et al., 2010; Hu et al., 2011), and the targeted genes were searched for using a 1-Mb sliding window. Only previously reported candidate genes located in these regions and registered in the HapMap (human) and the Bovine HapMap databases were incorporated into the analysis.
Results
Population stratification
In the initial GWAS analysis without adjusting for PCs of genetic variation, the distribution of the observed P-values shows evidence for systematic bias (Figure 2). The value of λGC is 2.323 (λGC=2.323). The systematic bias most likely resulted from confounding by the latent sub-structuring within the population (Li et al., 2011). After adjusting for the first four PCs of genetic variance, the majority of the residual confounding resulting from population stratification (λGC=1.04 for the adjusted analyses) was removed. Further analysis by adjusting 10 PCs gave a similar λGC value. Therefore, all of the association results reported below are based on the analyses adjusted for the first four PCs of genetic variance.

Quantile–quantile plot (Q–Q plot). Quantile–quantile plot of the −log10(P-value) from an analysis of the GWAS that did not adjust for principal components of genetic variation (black dots) and an analysis that adjusted for the four principal components (red dots).
Association study and syntenic mapping
The thresholds of genome-wide significance at empirical levels of 5 and 1% after Bonferroni adjustment were 1.057 × 10−6 and 2.11 × 10−7, respectively. An empirical significance level of 1% (nominal P<2.11 × 10−7=0.01/47303; see Table 1) was set in order to minimize the effect of relatively small sample size. We identified significant (genome-wide P<2.11 × 10−7) 35 SNPs associated with the variation among all the coat colours studied. Manhattan plot of −log10 (P-value) across the genome after the GC correction is shown in Figure 3. Of the 35 SNPs, 18 showed very significant evidence for association with coat colour when comparing the Finnsheep with white coat colour versus without (Table 1), and 25 were located in or adjacent to the five sheep coat colour-associated genes (ASIP, TYRP1, KIT, MC1R and MITF) reported previously (Table 1). In particular, our GWAS revealed multiple significant associations on chromosomes 13 (n=12), 19 (n=5) and 2 (n=4) at short intervals (<6 Mb for the associations on OAR13 and OAR19; <ca. 9 Mb for three associations on OAR2).

GWA scan for the coat colours and patterns of sheep. Negative log10 P-values by principal component analysis are shown for SNPs that passed quality control. The dashed line is the genome-wise significance threshold (P=2.11 × 10−7).
Syntenic mapping for the previously reported candidate genes in sheep genomic resources revealed that the significant (P<2.11 × 10−7) association for coat colour were located close to three previously known candidate genes (TYRP1, ASIP and MITF; see Supplementary Figure S1) for coat colours. All the three genes are within the intervals of the significant SNPs identified as predicated in the Real Sheep Genome Oar v3.1. The most significant SNP associations were on chromosome 19; the strongest signals were attained at OAR19_33531771.1 and s00651.1 bordered by MITF (P=6.9 × 10−12 at 33 531 717 bp and P=2.7 × 10−11 at 38 015 761 bp; see Table 1), when the analysis was done for coat colour from white to black/white spotted (that is, all the colours). These 2, and another 16, SNPs were consistently indicated as being significantly associated with coat colour in the following white versus non-white analysis. The SNP s66432.1 in ASIP showed the strongest signal of association with the coat variation between white and non-white. Genotype frequencies of the significant SNP s66432.1 in the Finnsheep population investigated were shown in a contingency table (Supplementary Table S2). However, we did not find significant associations with coat colour from grey to black/white spotted (that is, non-white colours). The combined P-value at the identified SNPs across the three analyses varied from 1.08 × 10−12 to 6.01 × 10−23 (Table 1), with P=7.95 × 10−22 at the SNP OAR19_33531771.1 and P=4.05 × 10−21 at the SNP s00651.1. However, notably, the significant SNPs identified were often located near (but not in) the genes. An additional careful inspection of genes flanking the significant SNPs and database searches of presumed function in humans and sheep did not detect any novel candidate genes that were associated with coat colour or other pigmentation-related traits.
We further inspected the SNP effects by looking at the coefficient of determination. The peak SNPs flanking the identified candidate genes explained on average ca. 31% of the phenotypic variance (from 20.3 to 37.1%). This is much higher than for the significant SNPs associated with hair colour and skin pigmentation in humans (for example, Han et al., 2008; Nan et al., 2009). We inspected genotypes of the most significant SNP s66432.1 in ASIP and found that 96% (48/50) of the Finnsheep is homozygous (A/A) in the white Finnsheep, but frequency of the homozygote (A/A) is extremely low (2%, 1/50) in the non-white Finnsheep. For the dominant or recessive model of the SNP s66432.1 in ASIP, the segregation of the F1 population gave a very good fit to a 3:1 ratio of white versus non-white (χ2=0.44; P=0.40–0.55), indicating a dominant effect of allele A to allele G. We also inspected genotypes of the peak SNPs neighbouring the three identified genes and found that most white Finnsheep were homozygous for the SNPs s29398.1 (G/G; 45/50, 90%) and s31905.1 (G/G; 46/50, 92%), whereas proportions of the two homozygotes were only 17.1% (7/41) for s29398.1 and 14.6% (6/41) for s31905.1 in the non-white Finnsheep.
SNPs and genes under selection
In the analysis of global FST for 47 303 SNPs at the genome-wide level, we detected the highest selection signal on OAR13 (Figure 4) between white and non-white individuals. The highest-ranked SNP (s66432.1; FST=0.731), which showed a single and strong selection signal, was located at the position 67.95 Mb within the ASIP gene. In sheep, the duplicated ASIP allele (s) was (were) found to account for the white and black sheep (for example, Norris and Whan, 2008; Fontanesi et al., 2011), and ASIP displayed strong evidence for positive selection in a quite recent study across a worldwide collection of breeds (Kijas et al., 2012).

Selection signals in the genome of the Finnsheep. Animals in white coat colour were compared with animals in non-white coat colour (that is grey, brown, black and black/white spotted). Pairwsie FST was calculated between the two groups of Finnsheep individuals for all 47 303 SNPs. A strong selection signal was observed on the chromosome 13 above the ASIP gene.
A total of four genomic regions containing ⩾9 consecutive significant average values <0.05 were identified to be under positive selection using the five-SNP sliding-window analysis (Table 2), whereas no region was observed with balancing selection. Signals of positive selection were identified spanning KIT, MITF, ASIP and TYRP1. The content of the genetically differentiated genomic regions will enrich the genes under directional selection, for example for the white coat colour given their roles in coat pigmentation in sheep.
Linkage disequilibrium
Pairwise tests of LD were performed with upstream and downstream regions neighbouring the significant SNPs included. There are 12 significant SNPs (4.2 × 10−11<P<1.3 × 10−7) mapping to the 5.9-Mb interval at OAR13. Three discrete haplotype blocks were identified in the 5.9-Mb peak of association (see Supplementary Figure S2). ASIP lies within one of the associated LD blocks that contains seven significant SNPs (s28243.1, s31905.1, s27858.1, OAR13_67687804.1, s13874.1, s66432.1 and s74265.1). Only two SNPs (s66432.1 and s12884.1) located in the inferred genomic region of ASIP (67.40–68.81 Mb; see Supplementary Figure S2) were genotyped in this study. Nevertheless, the significant SNPs in the various LD blocks did not show any strong LD with the two SNPs in ASIP. The estimated P-values of s12884.1 did not reach genome-wide significance (P>10−4, data not shown) when analysis was implemented across all coat colours and patterns, while marginally significant P-values (1.1 × 10−5 and 3.7 × 10−6, respectively; data not shown) were recorded in the non-white and white versus non-white analyses.
At OAR19, five significant SNPs (6.9 × 10−12<P<3.2 × 10−8) span 4.8 Mb, with two SNPs (OAR19_33531772.1 and s00651.1) showing the most extreme significant values (Table 1). Out of the five significant SNPs, two (OAR19_33278780.1 and OAR19_33531772.1) are located in one LD block, which is ∼790 kb and includes MITF in the genomic region of 33.14–3.19 Mb (see Supplementary Figure S2). Another LD block was of ∼680 kb, but there was no significant SNP within or around it (see Supplementary Figure S2). Another region showing significant association with sheep coat colour is on chromosome 2 (OAR2). The three significant SNPs (3.8 × 10−11<P<1.2 × 10−7) were located within a 12.14-Mb wide region. We identified four LD blocks, and none of the three significant SNPs occurred in them. The candidate gene TYRP1, covering a genomic region of ca. 20 kb from 85.49 to 85.51 Mb, is separated from the LD blocks (see Supplementary Figure S2). Unfortunately, no SNP within the candidate genes MITF and TYRP1 was genotyped in the study.
The s66432.1 SNP in the ASIP gene was highly associated with white versus non-white coat colour, but relatively weakly associated with other coat colour variations in the pooled analysis of all the colours (see Table 1). This SNP explained 2.3% of residual variation in coat colour from grey to black/white spotted (non-white coat colour), after controlling for the first four PCs. The index s66432.1 SNP is in significant LD (r2=0.321–0.814) with six other variants near the ASIP gene in the analysis of white versus non-white (Figure 5). We did not detect any significant LD when the analysis was limited to the non-white (from grey to black/white spotted) samples (Figure 5).

Regional plots of the 15 identified SNPs covering the ASIP in the analysis of (a): white versus non-white and (b): only the non-white Finnsheep. Genotyped SNPs passed quality control measures in GWAS plotted with the P-values (as –log10(P-value)) as a function of genomic position. The two SNPs genotyped in ASIP are represented by a diamond. Colour of the remaining SNP (circles) indicated LD with the two SNPs according to a scale from r2=0 to r2=1 based on pairwise r2 values.
Discussion
Pigmentation in domestic animals like sheep (including the wild population of Soay sheep) has been long considered to be tightly regulated by genetic variations in homologous genes identified in humans and model species (for example, Klungland et al., 2000; Andersson, 2001; Cockett et al., 2001; Sturm, 2006; Gratten et al., 2012). However, candidate gene approaches that are designed according to a priori hypotheses cannot evaluate the effect of the gene in the whole-genome context, nor can they identify other genes contributing to the phenotype. In addition, very few candidate genes with common genetic variants accounting for the variation in coat colour of sheep have been identified in earlier investigations. Therefore, by employing a hypothesis-free approach we conducted a GWAS and selection test of coat colours and patterns in an isolated sheep population, the Finnsheep breed in Finland. We identified a few new variants (see Table 1) associated with variation in coat colour among the Finnsheep individuals, and in particular one variant (s66432.1) in the ASIP gene that accounts for most of the sheep coat-colour variation between white and non-white forms.
Candidate genes associated with sheep coat colours
By carrying out a GWAS in a Finnsheep cohort, we identified up to 35 SNPs that are associated with variation among all the sheep coat colours at the genome-wide significance levels. Additionally, we detected genomic regions close to three previously identified pigmentation candidate genes (TYRP1, ASIP and MITF), and all three genes are involved in the classical pathways of melanogenesis. The three genomic regions explain ca. 22.1% of the residual variation in coat colour after correcting for the top four PCs of genetic variation, and the most substantial contribution (12.5%) was from the significant variants in and near the ASIP gene. The hypothesis-free study conducted reported significant SNPs in the candidate genes that could be predicted using a candidate gene approach, but additional SNPs outside the candidate genes were detected as well. These three genes were also found to be under strong selection pressure in a genome-wide selection test among sheep breeds of different coat colours (for example, Kijas et al., 2012). Thus, it seems likely that along with the variation in coat colours, the allele frequency distribution of variants identified was probably driven by positive selection at certain candidate genes or genomic regions.
ASIP as a candidate gene for white versus non-white
The ASIP gene encodes a secreted protein that affects the quality of hair pigmentation and has been shown to have a central role in neuroendocrine functions of melanocortin action (for example, Norris and Whan 2008). Previous studies have also identified the ASIP protein as a competitive antagonist of the melanocyte-associated melanocortin receptor (MC1R) gene, which inhibits the binding of melanocyte stimulating hormone (α-MSH) and subsequent induction of eumelanin synthesis (Lu et al., 1994; Parsons et al., 1999). Increased expression of MITF and its activation by phosphorylation stimulate the transcription of tyrosinase-related protein 1 (TYRP1). We detected significant LD between the variant s66432.1 and surrounding SNPs in the analysis of white versus non-white samples (Figure 5). This suggests that the signals detected on OAR13 might be due to the functional variant in the ASIP gene, which could be verified by further transcript and protein investigation. As we employed stringent significance thresholds for statistical significance, we were only able to detect common variants with large effects. However, some variants with relatively smaller effects are completely masked in the presence of others, and these variants remain to be identified. This might explain why we failed to detect genome-wide significance at the other SNP (s12884.1) in ASIP, which shows slightly lower significant values than the threshold. In addition, due to the small sample size used for the study, there is good reason to believe that a proportion of such low-frequency variants (for example, Speliotes et al., 2010) and short insertion–deletion polymorphisms (for example, Norris and Whan, 2008) were not captured. Other genes and variants could be identified if the sample size studied could be larger and the density of the Beadchip designed could be higher, as several other GWAS of skin or hair colour phenotype in human (for example, Nan et al., 2008, 2009).
The contribution of ASIP to sheep coat colour was investigated via classic genetics from the early 1920s. The dominant white/tan (AWt) allele of ASIP was found to account for the white-wool phenotype in modern domestic sheep, whereas the most recessive allele, non-Agouti (Aa), explained uniformly eumelanic (black/brown) wool (Adalsteinsson, 1970). The badgerface (Ab) allele of ASIP results in a pale dorsal pheomelanic and darker ventral eumelanic pattern; the allele is recessive to AWt and dominant to Aa. The ASIP gene has been well characterized in mice and humans (for example, Wilson et al., 1995; Klungland et al., 2000), and its Bos taurus cattle homologues were identified as the genomics of B. taurus developed (for example, see the review by Lauvergne et al. (1989)). Several recognized natural patterns and many novel mutations in the ASIP gene, such as SNPs, insertion–deletion and tandem gene duplication, have been identified in mammals (for example, cattle (Dreger, 2008), dogs (Kerns et al., 2004), horses (Rieder et al., 2001), pigs (Mao et al., 2001), macaques (Nakayama et al., 2010), alpacas (Feeley et al., 2011) and rabbit (Fontanesi et al., 2010)). These include both spontaneous and induced mutations. In domestic sheep breeds, it was recently reported that a duplication of the ASIP gene that affects its expression accounts for white and black sheep (Norris and Whan, 2008). Mutations in ASIP have also been shown to be responsible for the wild-/self-type coat pattern polymorphism in Soay sheep (Gratten et al., 2010). Also, the duplicated copy allele (s) in ASIP gene was found to be associated with grey coat colour (Fontanesi et al., 2011), which could explain the observation that coat-colour variation from grey to black/white spotted (non-white coat colour) was marginally explained by SNPs in and close to the ASIP gene. In addition, recessive genetic variants at the ASIP were found to be associated with reduced lifetime fitness in a wild population of Soay sheep (Gratten et al., 2012). The most likely explanation for the significant signal at s66432.1 SNP could be that mutation in the ASIP gene affects pigmentation by interfering in the αMSH–MC1R interaction, which, in turn, results in a pigment-type switch from eumelanins to pheomelanins. Nevertheless, until now, without the benefit of sheep whole-genomic data, elucidation of the molecular mechanisms of the ASIP gene regulating coat colour in domestic sheep would need additional efforts. Given that some colour and pattern alleles are known to be penetrant (for example, Eizirik et al., 2010), it would be also interesting to assess the dominant and recessive models of gene actions (for example, dominant and recessive epistasis; complementary or duplicate gene action) among the significant SNPs. However, due to the limited inheritance data among the individuals (for example, individuals from F2 and further generations) in this study (see Li et al., 2011), further genetic segregation investigations on the significant SNPs identified here are necessary for understanding the inheritance of the coat colour and pattern in sheep.
Selection signals at ASIP and the limitations
We detected the strongest selection signal in ASIP (Figure 4), a gene involved in melanocyte development across multiple mammals including sheep (see the reviews by Klungland et al. (2000) and Andersson and Georges (2004)). Earlier studies suggest that ASIP gene was targeted by man’s breeding desire for favourable and consistent coat pigmentation (that is, white) in the Finnsheep (see for example, Tapio et al., 2003; Li et al., 2011). A genome-wide selection test using global FST revealed four genomic regions with evidence for positive selection (Table 2), three of which containing the KIT, ASIP and MITF candidate genes were also detected in a previous genome-wide scan for selection signal across a worldwide collection of 74 sheep populations (Kijas et al., 2012). However, it is difficult to clarify the complex links between effect of genetic selection and the phenotypic consequence (see also Kijas et al., 2012), although convincing signals were detected at the SNP s66432.1 where selection demonstrated to have taken place driven by a developed breeding objective.
In the Finnsheep population, our study revealed strong divergent selection pressures at the ASIP gene in different coat colours. The ubiquity of white wool in domestic sheep could be a result of the universal human preference (see also Fang et al., 2009). One recent illustrative example of human preference for specific coat colour in domestic animals includes white colour in horses (Rosengren Pielberg et al., 2008). Utilization of the ASIP gene identified here will facilitate the marker-assisted breeding for specific coat colours in sheep.
While our approaches to identify relevant genetic variants under selection are effective, there are limitations in them. Firstly, our SNP data include only a fraction of the coding portion of the sheep genome, and higher density discovery and analysis would enable further genomic approaches to relevant genetic variation under selection. Moreover, the FST-outlier approach is restricted by a few underlying assumptions regarding to the demographic and mutational models (for example, genetic drift and island model), which might not exactly reflect the true demographic history of the Finnsheep population investigated in this study. Thus, application of multiple neutrality tests, which were based on different assumptions and parameter estimation, is a practical approach to confirm the candidate status of particular loci. Only loci identified as outliers by multiple methods are considered for subsequent validation investigations (see for example, Li et al., 2010). Finally, future approaches using sets of SNPs in target candidate genes, as opposed to genome scans, may be more fruitful in identifying loci associated with specific selection and adaptation scenarios (see the reviews by Slate et al. (2010) and Stapley et al. (2010)).
It is important to also note that the sample size was relatively small in this study. However, the results of the qualitative trait GWAS demonstrated that the sample size used was sufficient to detect a major genetic effect such as that manifested at the ASIP locus. As reported in earlier studies of population isolates (for example, Peltonen et al., 2000; Hill et al., 2010), the high levels of LD in the isolated Finnsheep population are a reflection of low effective population size, which enables detection of association with smaller sample sizes. However, in the analysis of only non-white colours that include less than half of the animals, the power of the test could be relatively low, and thus contributes partly to the lack of significant associations (see for example, Han et al., 2008; Hill et al., 2010). We also noticed that the sample size is different among the subpopulations of different coat colours, particularly only a single black/white spotted sheep. Nevertheless, given the similarly small proportion of black/white spotted sheep in the total Finnsheep breed (<1%; Puntila, 1997; Puntila et al., 1999), we think it should make more sense to include the single black/white spotted sheep in the analysis than to remove it. Another potential limitation exists in the fact that historically the development and application of genome-wide SNP tools in sheep has been later than those in other major livestock like cattle and swine. As a result this has limited the ability or possibility to identify or characterize novel target genes for specific traits of interest, including the coat colour and patterns investigated in this study. For example, due to the low-resolution syntenic maps of sheep genes to human and cattle, we cannot map SNPs near or in any genes and cannot locate a variant in the functional region (intron and exon) of a candidate gene.
Future directions and conclusions
In addition to providing new insights into the genetics and regulation of sheep coat colours and patterns, the study also raised several questions for future investigations. Sequencing of the candidate genes, ASIP in particular, in all the sheep used for the GWAS analysis would be an interesting study in the near future. This could reveal other possible causative mutation(s) behind the coat colour in the Finnsheep. As reported by Norris and Whan (2008), it could be also beneficial to genotype the duplicated ASIP alleles from the duplication 5′ breakpoint. Moreover, for the SNPs not mapped to known genes, it would be interesting to clarify if they are in the vicinity of other functional elements (for example, enhancers). In addition, SNP genotyping with dense array, gene fine mapping approaches and larger sample size with similar individuals for different colours and patterns will be necessarily needed to further characterize genetic variants at or near the identified genes and to support further investigations for the genes with smaller effect that were not identified in this study. As in a GWAS some true associations might be weakly associated with the phenotype studied, replication studies (or at least one additional replicated study) in a large scale is very necessary for confirming the earlier results, otherwise some of the true associations may be missed (for example, Hunter et al., 2007; Nan et al., 2009). Thus, further large-scale replicated investigations of the strong SNP associations and of the genomic regions containing TYRP1, MITF and ASIP genes in other independent cohorts or populations are necessary for confirmation and strengthening the credibility of our findings.
To the best of our knowledge, this study is the first GWAS on a pigmentation phenotype, an economically and ecologically (for example, the coat pattern is associated with reduced lifetime fitness in a wild population of sheep; Gratten et al., 2012) important trait in sheep. We provide strong evidence from a multi-stage study for TYRP1, MITF and ASIP genes being associated with sheep coat colours and patterns, and in particular identify a common genetic variant in ASIP accounting for coat-colour variation in white versus non-white. The associations near and within the ASIP gene and the known role of ASIP in melanogenesis together suggest an intriguing new insight into sheep coat pigmentation. This study therefore contributes to a long-term collective effort to identify valuable genes and mutations from the sheep genetic resources for future genetic breeding. Our study provides a novel entry point for developing genetic testing for sheep coat colours and patterns in animal breeding programmes. Last but not the least, the study has also yielded new insights into the evolution of complex phenotypic traits under artificial and/or natural selection in other domestic and wild animals.
Data archiving
Data deposited in the Dryad repository: doi:10.5061/dryad.fb6qg.
References
Adalsteinsson S . (1970). Colour inheritance in Icelandic sheep and relation between colour, fertility and fertilisation. J Agric Res Iceland 2: 3–135.
Andersson L . (2001). Genetic dissection of phenotypic diversity in farm animals. Nat Rev Genet 2: 130–138.
Andersson L, Georges M . (2004). Domestic-animal genomics: deciphering the genetics of complex traits. Nat Rev Genet 5: 202–212.
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G . (2008). LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9: 323.
Bacanu SA, Devlin B, Roeder K . (2002). Association studies for quantitative traits in structured populations. Genet Epidemiology 22: 78–93.
Beaumont MA, Balding DJ . (2004). Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol 13: 969–980.
Becker D, Tetens J, Brunner A, Bürstel D, Ganter M, Kijas J . (2010). Microphthalmia in Texel sheep is associated with a missense mutation in the paired-like homeodomain 3 (PITX3) gene. PLoS One 5: e8689.
Beraldi D, McRae AF, Gratten J, Slate J, Visscher PM, Pemberton JM . (2006). Development of a linkage map and mapping of phenotypic polymorphisms in a free-living population of Soay sheep (Ovis aries). Genetics 173: 1521–1537.
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K . (2009). Coat variation in the domestic dog is governed by variants in three genes. Science 326: 150–153.
Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Mainland I et al. (2009). Revealing the history of sheep domestication using retrovirus integrations. Science 324: 532–536.
Cockett NE, Shay TL, Smit M . (2001). Analysis of the sheep genome. Physiol Genomics 7: 69–78.
Costin GE, Hearing VJ . (2007). Human skin pigmentation: melanocytes modulate skin color in response to stress. FASEB J 21: 976–994.
Dalrymple BP . (2007). Using comparative genomics to reorder the human genome sequence into a virtual sheep genome. Genome Biol 8: R152.
Davenport CB . (1905). The origin of black sheep in the flock. Science 22: 674–675.
Deng W, Tan Y, Wang X, Xi D, He Y, Yang S et al. (2009). Molecular cloning, sequence characteristics, and polymorphism analyses of the tyrosinase-related protein 2/DOPAchrome tautomerase gene of black-boned sheep (Ovis aries). Genome 52: 1001–1011.
Devlin B, Roeder K . (1999). Genomic control for association studies. Biometrics 55: 997–1004.
Dreger DL . (2008). The genetics of variant red and changeling coat colour phenotypes in Holstein cattle. MSc thesis. Department of Animal and Poultry Science University of Saskatchewan: Saskatoon, SK, Canada.
Eizirik E, David VA, Buckley-Beason V, Roelke ME, Schäffer AA, Hannah SS et al. (2010). Defining and mapping mammalian coat pattern genes: multiple genomic regions implicated in domestic cat stripes and spots. Genetics 184: 267–275.
Feeley NL, Bottomley S, Munyard KA . (2011). Three novel mutations in ASIP associated with black fibre in alpacas (Vicugna pacos). J Agric Sci 149: 529–538.
Fontanesi L, Dall’Olio S, Beretti F, Portolano B, Russo V . (2011). Coat colours in the Massese sheep breed are associated with mutations in the agouti signalling protein (ASIP) and melanocortin 1 receptor (MC1R) genes. Animal 5: 8–17.
Fontanesi L, Forestier L, Allain D, Scotti E, Beretti F, Deretz-Picoulet S et al. (2010). Characterization of the rabbit agouti signaling protein (ASIP) gene: transcripts and phylogenetic analyses and identification of the causative mutation of the nonagouti black coat colour. Genomics 95: 166–175.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B et al. (2002). The structure of haplotype blocks in the human genome. Science 296: 2225–2229.
Gratten J, Pilkington JG, Brown EA, Beraldi D, Pemberton JM, Slate J . (2010). The genetic basis of recessive self-colour pattern in a wild sheep population. Heredity 104: 206–214.
Gratten J, Pilkington JG, Brown EA, Clutton-Brock TH, Pemberton JM, Slate J . (2012). Selection and microevolution of coat pattern are cryptic in a wild population of sheep. Mol Ecol 21: 2977–2990.
Han J, Kraft P, Nan H, Guo Q, Chen C, Qureshi A et al. (2008). A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet 5: e1000074.
Hill EW, McGivney BA, Gu J, Whiston R, MacHugh DE . (2010). A genome-wide SNP-association study confirms a sequence variant (g.66493737C>T) in the equine myostatin (MSTN) gene as the most powerful predictor of optimum racing distance for thoroughbred racehorses. BMC Genomics 11: 552.
Hu X, Pickering E, Liu YC, Hall S, Fournier H, Katz E et al. (2011). Meta-analysis for genome-wide association study identifies multiple variants at the BIN1 locus associated with late-onset Alzheimer’s disease. PLoS One 6: e16616.
Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE et al. (2007). A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 6: 870–874.
Jackson IJ . (1994). Molecular and developmental genetics of mouse coat colour. Annu Rev Genet 28: 187–217.
Johnston SE, McEwan JC, Pickering NK, Kijas JW, Beraldi D, Pilkington JG et al. (2011). Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol Ecol 20: 2555–2566.
Kayser M, Liu F, Janssens AC, Rivadeneira F, Lao O . (2008). Three-genome-wide association studies and a linkage analysis identify HER2 as a human Iris color gene. Am J Hum Genet 82: 411–423.
Kerns JA, Newton J, Berryere TG, Rubin EM, Cheng JF, Schmutz SM et al. (2004). Characterization of the dog Agouti gene and a nonagouti mutation in German Shepherd Dogs. Mamm Genome 15: 798–808.
Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M et al. (2012). Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol 10: e1001258.
Kijas JW, Townley D, Dalrymple BP, Heaton MP, Maddox JF, McGrath A et al. (2009). A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS One 3: e4668.
Klungland H, Olsen HG, Hassanane MS, Mahrous K, Våge DI . (2000). Coat colour genes in diversity studies. J Anim Breed Genet 117: 217–224.
Kristiansson K, Naukkarinen J, Peltonen L . (2008). Isolated populations and complex disease gene identification. Genome Biol 26: 109.
Lambrides CJ, Godwin ID, Lawn RJ, Imrie BC . (2004). Segregation distortion for seed testa color in Mungbean (Vigna radiata L. Wilcek). J Hered 95: 532–535.
Lauvergne JJ, Renieri C, Silvestrelli M, Valfré F . (1989). Genetic analysis of cattle coat color from crossbreds with Swiss Brown. J Anim Breed Genet 106: 389–397.
Li MH, Iso-Touru T, Laurén H, Kantanen J . (2010). A microsatellite-based analysis for the detection of selection on BTA1 and BTA20 in northern Eurasian cattle (Bos taurus) populations. Genet Sel Evol 42: 32.
Li MH, Merilä J . (2010). Extensive linkage disequilibrium in a wild bird population. Heredity 104: 600–610.
Li MH, Strandén I, Kantanen J . (2009). Genetic diversity and pedigree analysis of the Finnsheep breed. J Anim Sci 87: 1598–1605.
Li MH, Strandén I, Tiirikka T, Sevón-Aimonen ML, Kantanen J . (2011). A comparison of approaches to estimate the inbreeding coefficient and pairwise relatedness using genomic and pedigree data in a sheep population. PLoS One 6: e26256.
Loehr J, Worley K, Moe J, Carey J, Coltman DW . (2008). MC1R variants correlate with thinhorn sheep colour cline but not individual colour. Can J Zool 86: 147–150.
Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T et al. (1994). Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature 371: 799–802.
Mao H, Ren J, Ding N, Xiao S, Huang L . (2001). Genetic variation within coat color genes of MC1R and ASIP in Chinese brownish red Tibetan pigs. Anim Sci J 81: 630–634.
Mather K . (1963) The Measurement of Linkage in Heredity. Methunen: London, UK.
Miller JM, Poissant J, Kijas JW, Coltman DW, The International Sheep Genomics Consortium. (2011). A genome-wide set of SNPs detects population substructure and long range linkage disequilibrium in wild sheep. Mol Ecol Resour 11: 314–322.
Myers S, Bottolo L, Freeman C, McVean G, Donnelly P . (2007). A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324.
Nakayama K, Shotake T, Takeneka O, Ishida T . (2010). Variations in the coding region of the Agouti signaling protein gene do not explain Agouti/non-agouti phenotypes in Macaques. J Mammalian Evol 17: 211–214.
Nan H, Kraft P, Qureshi AA, Guo Q, Chen C, Hankinson SE et al. (2009). Genome-wide association study of tanning phenotypes in a population of European ancestry. J Invest Dermatol 129: 2250–2257.
Norris BJ, Whan VA . (2008). A gene duplication affecting expression of the ovine ASIP gene is responsible for white and black sheep. Genome Res 18: 1281–1293.
Parsons YM, Fleet MR, Cooper DW . (1999). The Agouti gene: a positional candidate for recessive self-colour black pigmentation in Australian Merino sheep. Aust J Agric Res 50: 1099–1103.
Patterson N, Price AL, Reich D . (2006). Population structure and eigenanalysis. PloS Genet 2: e190.
Peltonen L, Palotie A, Lange K . (2000). Use of population isolates for mapping complex traits. Nat Rev Genet 1: 182–190.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D . (2006). Principal component analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909.
Puntila M-L . (1997). Selection criteria and fleece characteristics for the Finnsheep. Newsletter Eur Fine Fibre Network 2: 5.
Puntila M-L, Nylander A, Nuutila E-M . (1999). Genetic parameters for wool traits in Finnsheep lambs. In: The 50th Annual Meeting of the European Association for Animal Production Book of Abstracts 5, p 264 Mimeogr. 8p. Zurich, Switzerland.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575.
Renieri C, Valbonesi A, La Manna V, Antonini M, Lauvergne JJ . (2008). Inheritance of coat colour in Merino sheep. Small Ruminant Res 74: 23–29.
Rieder S, Taourit S, Mariat D, Langlois B, Guerin G . (2001). Mutations in the agouti (ASIP), the extension (MC1R), and the brown (TYRP1) loci and their association to coat color phenotypes in horses (Equus caballus). Mamm Genome 12: 450–455.
Rosengren Pielberg G, Golovko A, Sundström E, Curik I, Lennartsson J, Seltenhammer MH et al. (2008). A cis-acting regulatory mutation causes premature hair graying and susceptibility to melanoma in the horse. Nat Genet 40: 1004–1009.
Scherag A, Jarick I, Grothe J, Biebermann H, Scherag S, Volckmar AL et al. (2010). Investigation of a genome wide association signal for obesity: synthetic association and haplotype analyses at the melanocortin 4 receptor gene locus. PLoS One 5: e13967.
Slate J, Santure AW, Feulner PG, Brown EA, Ball AD, Johnston SE et al. (2010). Genome mapping in intensively studied wild vertebrate populations. Trends Genet 26: 275–284.
Slominski A, Tobin DJ, Shibahara S, Wortsman J . (2004). Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev 84: 1155–1228.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU et al. (2010). Association analyses of 249 796 individuals reveal 18 new associated with body mass index. Nat Genet 42: 948.
Sponenberg DP . (1997). Genetics of colour and hair texture. In: Piper LR, Ruvinsky A, eds The Genetics of Sheep. CAB International: Wallingford, UK. pp. 51–85.
Stapley J, Reger J, Feulner PG, Smadja C, Galindo J, Ekblom R et al. (2010). Adaptation Genomics: the next generation. Trends Ecol Evol 25: 705–712.
Sturm PA . (2006). A golden age of human pigmentation genetics. Trends Genet 22: 464–468.
Sulem P, Gudbjartsson DF, Stacey SN, Helgason A, Rafnar T, Magnusson KP et al. (2007). Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat Genet 39: 1443–1452.
Tapio M, Miceikiené I, Vilkki J, Kantanen J . (2003). Comparison of microsatellite and blood protein diversity in sheep: inconsistencies in fragmented breeds. Mol Ecol 12: 2045–2056.
Våge DI, Fleet MR, Ponz R, Olsen RT, Monteagudo LV . (2003). Mapping and characterization of the dominant black colour locus in sheep. Pigment Cell Res 16: 693–697.
Wang N, Aksey JM, Zhang K, Chakraborty R, Jin L . (2002). Distribution of recombination crossover and the origin of haplotype blocks: the interplay of population history, recombination, and mutations. Am J Hum Genet 71: 1227–1234.
Warwick BL, Berry RO, Davis SP . (1957). Inheritance of recessive black in sheep. J Hered 48: 254–258.
Wickham H . (2009) Ggplot: Elegant Graphics for Data Analysis. Springer: New York, NY, USA.
Wilson BD, Ollmann MM, Kang L, Stoffel M, Bell GI, Barsh GS . (1995). Structure and function of ASP, the human homolog of the mouse agouti gene. Hum Mol Genet 4: 223–230.
Acknowledgements
The International Sheep Genomics Consortium (ISGC, http://www.sheephapmap.org) is acknowledged for creating the genomic data. We thank the 100-talent Program of the Chinese Academy of Sciences (M-HL) and the Academy of Finland (grants No. 250633 and No. 256077; M-HL) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Rights and permissions
About this article
Cite this article
Li, MH., Tiirikka, T. & Kantanen, J. A genome-wide scan study identifies a single nucleotide substitution in ASIP associated with white versus non-white coat-colour variation in sheep (Ovis aries). Heredity 112, 122–131 (2014). https://doi.org/10.1038/hdy.2013.83
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.83
Keywords
This article is cited by
-
Genetics of the phenotypic evolution in sheep: a molecular look at diversity-driving genes
Genetics Selection Evolution (2022)
-
Analysis on the desert adaptability of indigenous sheep in the southern edge of Taklimakan Desert
Scientific Reports (2022)
-
Unveiling genomic regions that underlie differences between Afec-Assaf sheep and its parental Awassi breed
Genetics Selection Evolution (2017)
-
Alteration of sheep coat color pattern by disruption of ASIP gene via CRISPR Cas9
Scientific Reports (2017)
-
Association of MITF loci with coat color spotting patterns in Ethiopian cattle
Genes & Genomics (2017)

{kind=link}
{kind=link}